HET ONTRAFELEN VAN DE
IMMUNOPATHOGENESE VAN DE
INFLAMMATOIRE MYOPATHIEËN:
DE DIFFERENTIËLE DIAGNOSE VAN LIMB-GIRDLE MUSCULAR
DYSTROPHY
Elise Velghe
Studentennummer: 01508964
Promotor: Dr. De Paepe Boel
Masterproef voorgelegd in het kader tot het behalen van de graad Master of Medicine in de Geneeskunde
Academiejaar: 2018 – 2020
Deze pagina is niet beschikbaar omdat ze persoonsgegevens bevat.
Universiteitsbibliotheek Gent, 2021.
This page is not available because it contains personal information.
Ghent University, Library, 2021.
Voorwoord/bedanking
Deze masterproef was voor mij de allerlaatste uitdaging in de opleiding geneeskunde voor ik mag beginnen aan de klinische stages. Om dit tot stand te brengen heb ik bijzonder veel hulp gekregen van een aantal mensen, die ik via deze weg graag zou bedanken.
Om te beginnen wil ik mijn promotor dr. Boel De Paepe bedanken voor de begeleiding de voorbije twee jaar. Ze nam de tijd om mijn vragen te beantwoorden, mijn werk grondig na te lezen, waardoor ik kon rekenen op een duidelijke feedback. Ze stuurde me bij waar nodig om tot een volwaardig resultaat te komen. Bijkomend heeft ze mij bijzonder veel bijgeleerd over het laboratoriumwerk binnen de dienst neuropathologie. Western blot en kleuringen werden mij getoond en voorzien van de nodige uitleg, zodat ik nadien op zelfstandige basis mijn eigen proeven kon afwerken.
Bijkomend geef ik graag een dankwoord aan Sophie D’hose en Caroline Merckx, aangezien ze in het laboratorium altijd paraat stonden om mijn vragen te beantwoorden en mij te helpen waar nodig met mijn experimenteel werk.
Vervolgens wil ik Prof. Dr. De Bleecker bedanken, omdat hij ervoor gezorgd heeft dat ik mijn klinisch aspect van mijn masterproef beter kon begrijpen. Het bijwonen van consultaties op de dienst neurologie, gaf mij een betere kijk op de ziekte, met name hoe deze zich manifesteerde bij de patiënten.
Tot slot verdienen mijn ouders ook een woord van dank. Bedankt voor de steun die jullie mij gegeven hebben gedurende mijn studies. Jullie hebben mij de kans gegeven om te groeien binnen deze opleiding, en er altijd voor mij te zijn ook wanneer het even minder ging.
Inhoudstafel
Abstract ... 1
Inleiding ... 2
1.
Conceptueel kader ... 2
2.
LGMD ... 3
2.1 Algemene beginselen ... 3 2.2 LGMD2A ... 5 2.2.1 Epidemiologie ... 5 2.2.2 Genetisch defect ... 6 2.2.3 Klinische presentatie ... 7 2.3 LGMD2I ... 9 2.3.1 Epidemiologie ... 9 2.3.2 Genetisch defect ... 9 2.3.3 Klinische presentatie ... 10 2.4 LGMD2J ... 11 2.4.1 Epidemiologie ... 11 2.4.2 Genetisch defect ... 11 2.4.3 Klinische presentatie ... 12 2.5 LGMD2L ... 12 2.5.1 Epidemiologie ... 12 2.5.2 Genetisch defect ... 12 2.5.3 Klinische presentatie ... 121.
Literatuuronderzoek... 14
2.
Spierbiopsie ... 14
2.1 Kenmerken van een goede biopsie ... 14
2.2 Histochemische kleuringen ... 14
2.2.1 Hematoxyline & eosine ... 15
2.2.2 PAS ... 15
2.2.3 Gomori trichoom ... 15
2.2.4 Mitochondriale kleuringen... 16
2.3 Immunohistochemische en –fluorescente kleuringen (IHC en IF) ... 17
2.4 Western blot ... 18
3.
Patiënten ... 21
Resultaten ... 22
1.
LGMD2A ... 22
1.1 Klinische en genetische karakteristieken ... 22
1.3 Immunofluorescente karakteristieken ... 24
1.4 Western blot ... 25
2.
LGMD2I ... 25
2.1 Klinische en genetische karakteristieken ... 25
2.2 Histologische karakteristieken ... 26
2.3 Immunofluorescente karakteristieken ... 27
2.4 Western blot ... 28
3.
LGMD2J ... 29
3.1 Klinische en genetische karakteristieken ... 29
3.2 Histologische karakteristieken ... 30
3.3 Immunofluorescente karakteristieken ... 31
3.4 Western blot ... 32
4.
LGMD2L ... 32
4.1 Klinische en genetische karakteristieken ... 32
4.2 Histologische karakteristieken ... 32 4.3 Immunofluorescente karakteristieken ... 33 4.4 Immunohistochemische karakteristieken ... 34 4.5 Western Blot. ... 34
5.
Inflammatoire kenmerken ... 35
Discussie ... 39
Referentielijst ... 49
Abstract
Inleiding Limb-Girdle Muscular Dystrophy (LGMD) is een heterogene groep erfelijke ziekten, gekenmerkt door een aantasting van de proximale spiergroepen ter hoogte van de bekkengordel. Autosomaal dominante en recessieve subtypes zijn geïdentificeerd, elk met hun eigen genetische, klinische en fenotypische karakteristieken.
Doelstelling Deze masterproef beschrijft de onderliggende immunopathogenese aan de hand van de myopathologische verschijnselen aanwezig in de spierbiopsie. Er wordt toegespitst op de differentiële diagnose met inflammatoire myopathieën en welke elementen cruciaal zijn om de diagnose te stellen.
Methode Op twee cohorten van respectievelijk acht en vijf patiënten met LGMD subtypes 2A, 2I, 2J en 2L, worden western blot (WB), celltellingen en (immuno)histologische, immunofluorescente kleuringen (IF) uitgevoerd om de voornaamste pathologische veranderingen in detail te beschrijven.
Resultaten LGMD2A (P1-2): fibrose, type I predominantie, atrofie, interne nuclei zijn kenmerkend . Dystrofine 1 en 3 (DYS1,3), alfa-dystroglycan (DAG1) zijn zwak bij beide patiënten, . Calpaïne 3 is zwak bij P2 op WB, MHC-I en MAC zijn positief op IF. LGMD2I (P3-4): Necrotische vezels en interne nuclei met lege halo zijn kenmerkend voor P3, terwijl P4 vooral lichte atrofie vertoont, en veel interne nuclei. Bij P3 zijn MHC-I, MAC en NCAM positief op IF, daar waar DAG1 verzwakt is. Bij P4 is voornamelijk de zwakke kleuring bij DAG1 en de MHC-I en MAC positiviteit kenmerkend. LGMD2J (P5-6): COX-deficiëntie wordt gevonden bij P5, terwijl bij P6 extensieve fibrose en predominantie type I op de voorgrond staat P6 is zwak voor DYS1,3 en DYSF op IF, terwijl CAPN3 afwezig is op WB. MHC-I en MAC zijn negatief, terwijl NCAM positief is op IF. LGMD2L (P7-8): P7 vertoont voornamelijk veel fibrose en necrose, terwijl P8 over het algemeen weinig histologische veranderingen vertoont. DYSF en SCG zijn zwak op IF, alsook CAPN3 op WB. Daarnaast vertoont P7 NCAM positiviteit. Bij P8 is MHC-I positief op IF. Bij de extra cohorte met LGMD2L patiënten is de WB voor ANO5 afwijkend bij 2 van de 3 patiënten. De IF-kleuring voor ANO5 vertoont geen verschil tussen de patiënten en controles.
Conclusie In het algemeen vertoont de aantasting van het spierbiopt sterke variatie, ook tussen patiënten van hetzelfde type. Aspecifieke histologische veranderingen zijn aanwezig bij alle patiënten, die myopathie kenmerken. Immunodetectie pikt veranderingen in verschillende dystrofine-geassocieerde eiwitten op, die wijzen in de richting van musculaire dystrofie, doch vaak secundair zijn aan het onderliggende gendefect. Inflammatoire kenmerken zijn prominent aanwezig, en bemoeilijken de differentiatie met inflammatoire myopathieën. ANO5 western blot kon slechts een deel van de LGMD2L patiënten ïdentificeren.
De beschrijving van de immunopathologische veranderingen in het spierweefsel kan het gevonden onderliggend gendefect verder ondersteunen. Genetische karakterisatie is van cruciaal belang, aangezien het moleculaire therapie in de toekomst mogelijk maakt. Verdere studies naar modifiers die de fenotypische variaties beter kunnen verklaren, zijn wenselijk alsook de herziening van de classificatie van de LGMD.
Inleiding
1. Conceptueel kader
Neuromusculaire aandoeningen vormen een breed scala van uiteenlopende ziekten, waarbij de skeletspieren ontoereikend functioneren, met een belangrijke impact op vele lichaamsfuncties. Aan de basis hiervan ligt een pathologie ter hoogte van de spiercellen zelf, maar eveneens kan het probleem zich situeren op het niveau van de aansturing, meer bepaald de bezenuwing (1).
De invloed van deze aandoeningen op het dagdagelijks functioneren van patiënten, brengt met zich mee dat een correcte diagnose van cruciaal belang is met het oog op het reduceren van morbiditeit en mortaliteit. Dit is op zich een moeilijke opdracht, aangezien vele ziekten slechts gekenmerkt worden door enkele vage symptomen met een belangrijke overlap gaande van moeheid, spierzwakte, over stijfheid tot hevige pijn. Omwille van die redenen is het van belang om niet alleen rekening te houden met de huidige klinische presentatie van de patiënt, maar eveneens om hun voorgeschiedenis mee in rekening te brengen. Binnen de subgroep van kinderen kan gesteld worden dat er eerder problemen zijn met de normale motorische eindpunten zoals lopen, wandelen en zitten. In de perinatale periode kunnen achterstand van foetale bewegingen, alsook polyhydramnios vroege tekenen zijn die eventueel kunnen wijzen in de richting van een neuromusculaire pathologie (1).
Naargelang de snelheid van de totstandkoming van symptomen, kan een onderscheid gemaakt worden in etiologie. Betreft het een abrupte onset, dan zal de oorzaak zich voornamelijk situeren binnen het kader van toxisch metabole aandoeningen. Duurt het daarentegen dagen tot weken vooraleer de patiënt in kwestie last ondervindt, dan zal er eerder sprake zijn van een inflammatoir/infectieus proces. Tenslotte onderscheiden we nog een genetische, degeneratieve basis, als de klachten pas na maanden tot jaren verschijnen (1). De laatstgenoemde categorie zal binnen dit onderzoek bijzondere aandacht verdienen, waarbij voornamelijk zal gewerkt worden rond spierdystrofie. Dit betreft een heterogene groep van erfelijke musculaire aandoeningen, waarbij de spierkracht geleidelijk aan afneemt door atrofie van de verschillende spieren (1-3). Het primair pathologisch mechanisme achter vele van deze
ziekten betreft een verlies van connectie tussen de extracellulaire matrix en het actine cytoskelet. Er bestaan verscheidene subtypes elk met hun eigen ziektefenotype, genetische achtergrond en epidemiologie (1).
Een voorbeeld uit dat brede scala is de spierdystrofie van Duchenne, een aandoening die gekenmerkt wordt door een gendefect, meestal een deletie of duplicatie op het X-chromosoom, waardoor voornamelijk jongens reeds op vroege leeftijd deze ziekte ontwikkelen (2-3). Het genetisch defect leidt tot een verlies van het eiwit dystrofine, een cytoskeletaal proteïne dat instaat voor sterkte, stabiliteit en functionaliteit van de myofibrillen in spieren. Het effect van een niet functionerend dystrofine is progressieve musculaire zwakte, inclusief respiratoire en cardiale betrokkenheid. Het klinische fenotype verschilt van persoon tot persoon, maar patiënten overlijden vaak vanaf de leeftijd van 25 jaar door een gecompromitteerde hart- en longfunctie (2).
Dit genetisch defect betreft slechts één van de vele subtypes die horen binnen de term spierdystrofie. Zo bestaat er eveneens het nosologische concept van LGMD, wat nu reeds 50 jaar oud is en staat voor Limb-Girdle Muscular Dystrophy of in de volksmond bekkengordeldystrofie. Deze term is tot stand gekomen door het feit dat verschillende auteurs concluderen dat bepaalde patiënten een overwegende zwakte vertonen met proximale distributie die niet X-chromosomaal overerfbaar is (1,5). Met het oog op behandeling en prognose is het van belang om duidelijk uit te kunnen maken welke personen zich al dan niet binnen deze categorie bevinden, rekening houdend met de enorme overlap die soms kan optreden met andere aandoeningen.
Voor het verdere verloop van deze masterproef is het noodzakelijk om een achtergrond omtrent LGMD te schetsen met inbegrip van epidemiologie, klinische en genetische karakteristieken
2. LGMD
2.1 Algemene beginselen
Limb-Girdle Muscular Dystrophy, bevat meer dan 30 verschillende types, en werd voor het eerst beschreven door Walton en Nattrass. Wanneer de eerste genetische loci voor LGMD ontdekt werden, kwam al snel het besef dat dit een genetisch heterogene groep was, presenterend met een predominantie van proximale spierzwakte (1,4-5). De distale spieren worden grotendeels gespaard, alsook is er een exclusie van de extra-oculaire spieren. De faciale aantasting is nooit aanwezig in de vroege stadia, maar kan wel optreden bij rolstoelgebonden patiënten met een veralgemeende spierzwakte (5). De manifestaties kunnen
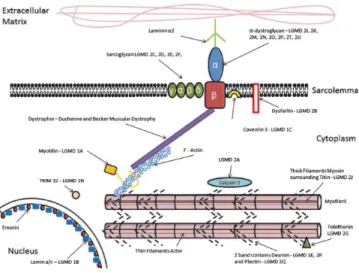
Figuur 1: Een weergave van de organisatie van het sarcolemma en de verschillende LGMD-subtypes geassocieerd met een defect in een welbepaald eiwit.
tot stand komen op elke leeftijd, inclusief vroege kindertijd, maar er is geen sprake van een congenitale afwijking. De aandoening wordt ernstiger na verloop van de jaren, maar de snelheid hiervan varieert naargelang het subtype waartoe de patiënt behoort (1).
De fenotypische presentatie is over het algemeen niet specifiek met als gevolg dat er geen verenigbaar biochemisch of pathofysiologisch concept is voor de gehele groep van LGMD. Daarom is het van belang om andere musculaire aandoeningen uit te sluiten. Een belangrijke differentiële diagnose dient gemaakt te worden met de idiopathische inflammatoire myopathieën, een groep van auto-immune ziekten, die gekarakteriseerd worden door de inflammatie van spieren en andere orgaansystemen. Poly- en dermatomyositis zijn de twee voornaamste categorieën binnen deze aandoening (1,5).
Wat uit de literatuur naar voor komt, is dat er een onderscheid gemaakt wordt op genetische basis, aangezien een klinische indeling niet mogelijk is door het brede spectrum dat zich binnen LGDM kan afspelen. De huidige indeling, maakt een onderscheid in twee grote groepen naargelang er een autosomale dominante (LGMD1) dan wel recessieve overerving (LGMD2) aanwezig is. De dominante vormen zijn milder en vertegenwoordigen slechts 10% van alle LGMD-patiënten (3,6). De autosomaal recessieve vormen, komen meer voor met een cumulatieve prevalentie van 1 op 150.000, met verschillen tussen de landen omwille van de distributie van het dragerschap en de graad van consanguiniteit (3). Binnen elke categorie wordt er gewerkt met subtypes refererend naar de genen en proteïnen die betrokken zijn in het pathologische proces. Over het algemeen kan gesteld worden dat de afwijkende eiwitten geassocieerd zijn met het sarcolemma, het contractiele apparaat, alsook proteïnen betrokken bij de glycosylatie of een enzymatisch proces (figuur 11) (1).
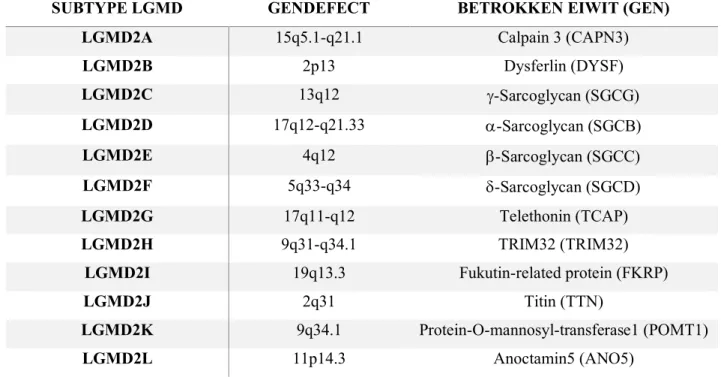
Tabel 1: De twaalf eerst gekarakteriseerde recessieve subtypes van LGMD2A tot 2L met hun bijhorend gendefect en betrokken eiwit.
Bovenstaande tabel toont de recessieve subtypes van LGMD, aangezien de geselecteerde cohorte binnen dit onderzoek uitsluitend type 2 patiënten betrof (1). De opsomming toont geen volledig beeld van deze categorie, omwille van het feit dat er reeds klassen zouden geïdentificeerd zijn gaande tot en met LGMD2Z.
De twee cohorten van respectievelijk acht (tabel 3) en vier (tabel 4) patiënten opgenomen in deze masterproef werden gediagnosticeerd met LGMD types 2A, 2I, 2J en 2L. Enkel deze types zullen daarom in de volgende hoofdstukken uitgebreid besproken worden.
2.2 LGMD2A
2.2.1 Epidemiologie
Deze autosomaal recessieve vorm van LGMD is de meest frequent voorkomende subgroep met een prevalentie in Europa van 1-9 op 100.000 (6-7). Over het algemeen kan gesteld worden dat het een bekende genetische entiteit is bij grote families op het Reunion eiland in de Indische Oceaan, langs de kust van Afrika, wellicht omwille van de aanwezigheid van consanguiniteit of bloedverwantschap, wat een verhoogd risico inhoudt op overerving van recessieve aandoeningen (1,6). Hier kan een prevalentie van 48 op 1.000.000 opgetekend worden (1).
SUBTYPE LGMD GENDEFECT BETROKKEN EIWIT (GEN)
LGMD2A 15q5.1-q21.1 Calpain 3 (CAPN3)
LGMD2B 2p13 Dysferlin (DYSF) LGMD2C 13q12 -Sarcoglycan (SGCG) LGMD2D 17q12-q21.33 -Sarcoglycan (SGCB) LGMD2E 4q12 -Sarcoglycan (SGCC) LGMD2F 5q33-q34 -Sarcoglycan (SGCD) LGMD2G 17q11-q12 Telethonin (TCAP) LGMD2H 9q31-q34.1 TRIM32 (TRIM32)
LGMD2I 19q13.3 Fukutin-related protein (FKRP)
LGMD2J 2q31 Titin (TTN)
LGMD2K 9q34.1 Protein-O-mannosyl-transferase1 (POMT1)
2.2.2 Genetisch defect
Meer dan 465 mutaties (missense, nonsense, deleties, inserties) zijn beschreven die zich bevinden op het niveau van de lange arm van chromosoom 15, meer bepaald ter hoogte van het CAPN3 gen, dat codeert voor het calcium-afhankelijk neutraal protease calpaïne 3 (1,6,8). Genotype en fenotype correlatie is niet 100% consistent, maar toch kan gesteld worden dat nonsense mutaties geassocieerd zijn met een ernstigere klinische presentatie, daar ze door incorporatie van een stopcodon leiden tot een volledige afwezigheid van het eiwit. Dit betekent op klinisch vlak dat de patiënten reeds voor de leeftijd van 15 jaar symptomen ondervinden en doorgaans nood hebben aan een rolstoel voor hun 40ste levensjaar. De effecten van missense mutaties daarentegen zijn bijzonder onvoorspelbaar op niveau van het fenotype (1).
Dit enzyme van 94kDa bevindt zich zowel in het cytosol als ter hoogte van de nucleus, waardoor er verondersteld wordt dat het een belangrijke rol speelt in de controle van specifieke transcriptiefactoren en in de regulatie van de differentiatie van spiercellen (1,3,6). Een functie van het calpaïne 3 eiwit is onder andere de inactivatie van IκBα, waardoor de nucleaire lokalisatie van NF-κB bevorderd wordt, wat op zijn beurt zal leiden tot activatie van anti-apoptotische genen. Bij LGMD2A, zien we dat er op basis hiervan een verhoogde anti-apoptotische celdood plaatsvindt van spiercellen (1,3,8).
Het IS2-domein, onderdeel van calpaïne 3 is een plaats van associatie met titine. Deze bevinding zou een functie kunnen aangeven voor CAPN3 in de controle van sacromere eiwitten door middel van remodelling of zou zelf gereguleerd kunnen worden via de binding aan titine. Dit laatstgenoemde eiwit kan gemuteerd zijn bij patiënten met LGMD2J, waarbij de fout zich dicht bij de calpaïne 3 binding bevindt (1).
Bijkomend is CAPN3 betrokken bij de regulatie van de protease activiteit in spieren. Een gebrek hiervan zou kunnen leiden tot een overactiviteit van andere proteasen met een afbraak van eiwitten tot gevolg (1). Er kan gesteld worden dat een deficiëntie van CAPN3 geassocieerd is met oxidatieve schade, Ca2+ dysregulatie, sarcomeer disorganisatie, mitochondriale abnormaliteiten, evenals onvoldoende aanpassing van spieren aan fysieke activiteit (afwijkende spieradaptatie) en toegenomen regeneratie van spierweefsel (figuur 2) (7).
2.2.3 Klinische presentatie
De aanvang van de ziekte is voornamelijk gesitueerd rond de leeftijd van 10 jaar, maar toch is een beduidend grote range zichtbaar gaande van 2 tot 40 jaar (1,6,9). Het gebruik van een rolstoel wordt noodzakelijk tijdens de verdere evolutie van de ziekte en patiënten worden over het algemeen rolstoelgebonden ongeveer 10 tot 20 jaar na de aanvang van de ziekte (6-8). Kijkend naar de prognose, kan gesteld worden dat de levensverwachting quasi gelijk is aan die van de gezonde populatie, mogelijks omdat, ondanks een uitgebreide distributie van calpaïne 3, het hart en de ademhalingsspieren slechts zelden betrokken zijn bij het ziekteproces (1,8). Helle Petri geeft aan dat er wel sprake kan zijn van aritmie en hypertrofe cardiomyopathie bij patiënten met LGMD2A, hoewel dit eerder een zeldzaam fenomeen is 10. Een gedaalde vitale capaciteit is bij sommigen merkbaar, alsook een stijging van het CK (creatine kinase) wat vrijkomt bij beschadiging van spierweefsel. De normale waarde is < 174 IU/l maar bij LGMD2A kan dit tot 50 keer verhoogd zijn (1,6-9).
Wanneer kinderen reeds op vroege leeftijd met deze ziekte te maken krijgen, worden de vroege motorische eindpunten over het algemeen wel gehaald. Het verschil zal zich vaak pas op latere leeftijd manifesteren, waarbij deze patiënten minder sterk zullen zijn in vergelijking met hun leeftijdsgenoten (1).
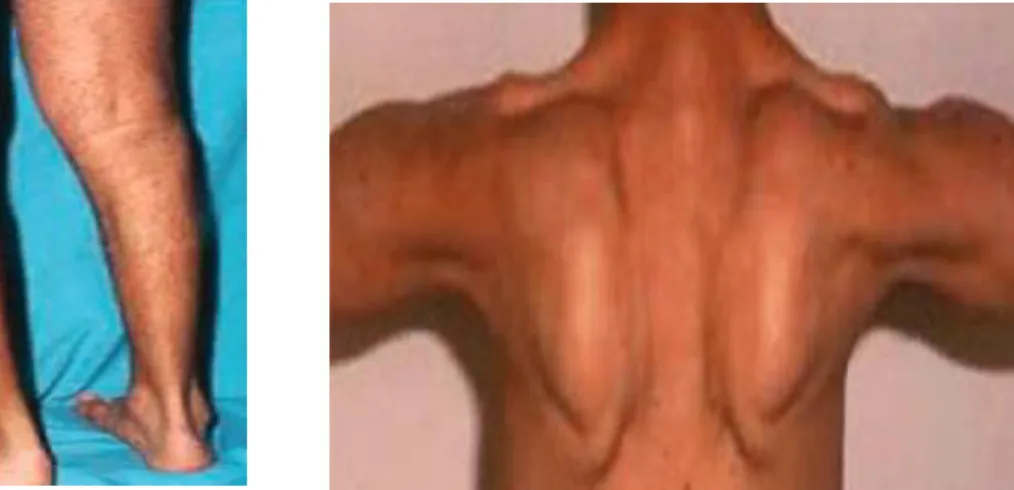
Bij de volwassen populatie, die hoofdzakelijk klaagt over spierstijfheid en myalgie bij inspanningen, is er een scapulohumerale pelvische distributie van spierzwakte met atrofie (figuur 32), waardoor de patiënten problemen ondervinden met trappen lopen alsook met snel
wandelen. De zwakte situeert zich ter hoogte van het bekken grotendeels op het niveau van de gluteus maximus, adductoren, flexoren en hamstrings, met een relatieve preservatie van de abductoren en distale spieren (1,9).
2
Belangrijk om te vermelden is dat er bij de evolutie van de ziekte een aantasting kan optreden van de tibiale spieren. Ter hoogte van de armen is er over het algemeen een betrokkenheid van de latissimus dorsi, rhomboideus, serratus anterior, pectoralis major met al dan niet symptomatische scapula alata (figuur 43). Dit betekent dat het schouderblad als het ware los
komt te staan van de thorax (1).
Bijkomend is er een relatief behoud van de triceps en blijven ook de nekflexoren gespaard. Bij dit type is er wel een laxiteit van de abdominale spieren aanwezig, waardoor een lumbale lordose zich eventueel kan manifesteren. Contracturen met voornamelijk een predominantie ter hoogte van de achillespees (figuur 34), elleboog en nek, kunnen zich ook presenteren bij
deze patiënten (1,9). Diepe peesreflexen ter hoogte van de onderste ledementen kunnen afwijkend zijn (1).
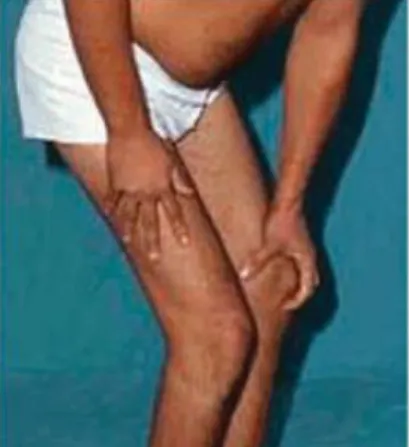
Een veel gebruikt onderzoek om spierzwakte van de onderste ledematen na te gaan is het zogenaamde Gower’s sign (Figuur 55). Deze test kan gebruikt worden bij alle patiënten met
een gekende proximale spierzwakte. De test is positief wanneer de persoon zich moet afduwen met de handen op de bovenbenen om vanuit zit recht te komen 1.
3/4
https://www.researchgate.net/figure/Clinical-features-frequently-observed-in-patients-with-LGMD-Gowers-manoeuvre-in-LGMD2A_fig1_319189482
5h ttps://www.researchgate.net/figure/Clinical-features-frequently-observed-in-patients-with-LGMD-Gowers-manoeuvre-in-LGMD2A_fig1_319189482
Figuur 3: Atrofie van musculus gastrocnemius met bijhorend een contracuur van de achillespees bij een patiënt met LGMD2A
2.3 LGMD2I
2.3.1 Epidemiologie
De relatieve proportie van de verscheidene subtypes verschilt tussen de onderzochte landen en tussen de verschillende artikels. Een artikel geeft aan dat in Engeland het grootste percentage bereikt wordt en dat het één van de meer voorkomende types is in Denemarken, Brazilië en de Verenigde Staten (11).
2.3.2 Genetisch defect
Het defect situeert zich op de lange arm van chromosoom 19, meer bepaald 19q13.3 waarbij er verscheidene mutaties gevonden worden in het Fukutin-related protein (FKRP) gen, dat nagenoeg 1,5 kilobase groot is (1,12-13). Dit leidt niet alleen tot LGMD2I, maar kan eveneens verantwoordelijk zijn voor de meer ernstige congenitale musculaire dystrofieën 1C (MDC1C) met secundair een laminin deficiëntie, alsook voor intermediaire fenotypes. Naargelang het soort en de grootte van het gendefect, is er een breedspectrum aan klinische fenotypes met variatie in ernst en tijdstip van diagnose. Het zijn vooral de missense en nonsense mutaties die geïdentificeerd worden, waarbij de 826C>A verandering bij de overgrote meerderheid van de LGMD2I patiënten wordt aangetoond (1,11-14).
FKRP zou een type II transmembranair eiwit zijn, dat gelijkenissen in sequentie vertoont met het glycosyltransferase (12). De functie is tot op van vandaag nog niet volledig opgehelderd, maar het zou voornamelijk een bijdrage hebben in de -dystroglycan O-glycosylatie ter hoogte van het Golgi apparaat. Het stabiliseert de link tussen het contractiele apparaat, meer bepaald tussen laminin 2 en de extracellulaire matrix (14). Het dystroglycan complex is belangrijk in de weefselopbouw en het behoud van adhesie (12).
Mutaties die voornamelijk leiden tot een defect in de translocatie van FKRP vanuit het endoplasmatisch reticulum naar het Golgi-apparaat, leiden tot de meer ernstige fenotypes,
Figuur 5: Gower's sign bij een patiënt met LGDM2A: om vanuit zit recht te komen duwt de patiënt zich af op de bovenbenen
Figuur 6 (links): Hypertrofie van de kuiten bij een patiënt met LGMD2I (13)
Figuur 7 (rechts): MRI met een vettige infiltratie van de vastus en biceps femoris bij een patiënt met LGMD2I (13)
gekend als het congenitale musculaire dystrofie 1C. De mutaties ter hoogte van het Golgi-apparaat zelf zijn eerder gecorreleerd met een mildere klinische presentatie, wat bekend staat als LGMD2I (12).
2.3.3 Klinische presentatie
Het ziektefenotype van LGMD2I is variabel in ernst, waarbij het mogelijk is dat de patiënt zich presenteert met een ernst gelijkaardig aan de ziekte van Duchenne. Dit betekent dat het ambulant functioneren reeds verdwijnt op het einde van de eerste of begin van de tweede decade. Over het algemeen is er een uitgebreide range gaande van 2 tot 40 jaar met een gemiddelde rond de 19 wat betreft de eerste symptomen. De fenotypische ernst van de klinische presentatie kan variëren, maar zij die reeds op vroege leeftijd met de aandoening te maken krijgen, hebben vertraagde motorische ontwikkeling, alsook hypotonie gedurende de eerste levensjaren. De gang kan abnormaal zijn en patiënten kunnen krampen en myalgie ondervinden reeds vanaf hun kindertijd (1,13).
De distributie van zwakte, al dan niet symmetrisch, situeert zich voornamelijk ter hoogte van de heup en bekkengordel. De flexoren van de nek en de axiale spieren zijn zwak, maar er wordt over het algemeen geen scapula alata vastgesteld. De biceps is beduidend sterker dan de triceps, alsook zien we dat de adductie van de schouder gemakkelijker verloopt dan de abductie. Ter hoogte van de onderste ledematen is er een verminderde plantaire flexie en is de quadriceps zwakker dan de hamstring. Eveneens kan aangetoond worden dat de extensie van de heup beduidend beter is dan de flexie, adductie en abductie. Contracturen worden niet vastgesteld, wel een lumbale lordose zonder tekenen van spinale rigiditeit (13). In vergelijking met LGMD2A, kan hier eventueel wel sprake zijn van hypertrofie van de kuit (figuur 6) omwille van het feit dat spierweefsel vervangen wordt door bindweefsel. In zeldzame gevallen kan er ook hypertrofie voorkomen van de tong (1,13).
Een bloedonderzoek toont een extreem verhoogde CK-waarde, variërend tussen de 750 en 10 000 IU/L, daar waar de normale waarde zoals reeds vermeld, gelegen is onder de 174 IU/L 13. Bij dit subtype kan er sprake zijn van myoglobinurie na inspanning (5). Wanneer eventueel een bijkomende MRI-scan uitgevoerd wordt, kunnen infiltraten van vet aanwezig zijn ter hoogte van de proximale onderste ledematen (figuur 7) (1,13).
Waar er bij LGMD2A slechts een matige aantasting is van het hart en de ademhalingsspieren, kunnen patiënten binnen deze 2I categorie zich presenteren met nachtelijke hypoventilatie door zwakte van het diafragma. Linker hartfalen met gedilateerde cardiomyopahtie kan eveneens voorkomen op oudere leeftijd, waarbij een daling van de ejectiefractie met meer dan 50% mogelijk is (10,12). Er zouden aanwijzingen zijn voor een daling van de ventriculaire ejectiefractie met 0,4% elk jaar (5).
2.4 LGMD2J
2.4.1 Epidemiologie
Deze vorm van LGMD is voor het eerst beschreven in Finland bij patiënten, waarbij er sprake is van een homozygote mutatie in het titine gen, welke in heterozygote vorm de zogenaamde tibiale musculaire dystrofie veroorzaakt (6).
2.4.2 Genetisch defect
Het titine gen heeft 363 exons coderend voor een groot structureel proteïne van het sarcomeer. Mutaties in het C-terminaal domein van M10 van titine veroorzaakt twee musculaire ziekten. Tibiale musculaire dystrofie is een dominante distale myopathie, veroorzaakt door een insertie of deletie in desbetreffende regio. In homozygoten, dezelfde mutatie kan LGMD2J veroorzaken met een vroegere onset en ernstigere fenotypische presentatie (1).
Het enzym titine vervult een waaier aan functies en fungeert als een continu filamentsysteem in de myofibrillen. Het zorgt namelijk voor het behoud van elasticiteit en morfologie van het sarcomeer tijdens spiercontractie, maar eveneens zorgt het voor de regulatie van de Z-schijf grootte. Het controleert de mechanische status van het sarcomeer door multiple regulatoren en kan informatie overbrengen naar signaalwegen die de spierfunctie regelen (1,15). Daarenboven fosforyleert dit eiwit telethonine, een proteïne dat deficiënt is in LGMD2G, alsook kan het zoals reeds vermeld calpaïne 3 binden, wat op zijn beurt kan leiden tot een secundaire deficiëntie ervan (1,6).
2.4.3 Klinische presentatie
De eerste tekenen van de ziekte komen meestal tot uiting in de eerste twee levensjaren, maar eveneens kan het zich pas manifesteren vanaf tienerleeftijd. De progressie vindt plaats over een periode van 20 jaar, waarna de patiënt rolstoel afhankelijk wordt (1,6,15).
De presentatie kenmerkt zich door regelmatig vallen, gedaalde inspanningstolerantie, moeite met lopen en het bestijgen van trappen, maar de cardiale functie is over het algemeen normaal (1,5,9,15).
2.5 LGMD2L
2.5.1 Epidemiologie
LGMD2L is over het algemeen het derde meest voorkomende subtype in Noord- en Centraal-Europa, waarbij aangegeven wordt dat het dubbel zo frequent voorkomt als het type LGMD2B (16).
2.5.2 Genetisch defect
Tal van causale mutaties zijn binnen deze aandoening beschreven in de literatuur, maar 60% ervan bevinden zich in het 5’ einde van het ANO5 gen. De duplicatie van 191 A is verantwoordelijk voor de meerderheid der gevallen, zowel in homozygote als compound heterozygote vorm. Het eiwit anoctamine 5, aanwezig in skeletspiercellen en schildklier, functioneert als een Ca2+ kanaal, maar ook als fosfolipide scramblase. Dit scramblase zorgt voor de translocatie van fosfolipiden binnenin de twee lagen van het celmembraan in respons op Ca2+ (1).
2.5.3 Klinische presentatie
LGMD2L wordt gekarakteriseerd door een aanvang op volwassen leeftijd, gemiddeld rond het 35ste levensjaar (17). De overgrote meerderheid van de patiënten ervaren moeite met
wandelen en dit voornamelijk bij grotere afstanden. Bijkomend zijn er klachten bij het bestijgen van de trap en bij het lopen op de tenen. Belangrijk is echter te vermelden dat sommige personen, voornamelijk vrouwen, zich uitsluitend presenteren met een verminderde sportprestatie, myalgie of spierstijfheid bij aanvang van de ziekte (1).
Een observatie van de spieren toont aan dat er proximaal een typisch asymmetrische zwakte aanwezig is ter hoogte van de heup. Het kan mogelijk zijn dat de distale spieren, met
voornamelijk de flexoren van knie en voet, betrokken zijn bij het proces met eventueel een hyperextensie van de knie (figuur 8) (17).
Ter hoogte van de schoudergordel, is de zwakte, die over het algemeen progressief is, voornamelijk te zien in de biceps, brachioradialis en triceps. Eveneens kan net zoals bij LGMD2A scapula alata vastgesteld worden. Er is een volledige preservatie van de handspieren. Atrofie wordt geobserveerd in de quadriceps femoris, hamstrings, mediale gastrocnemius, biceps en brachioradialis. Het laterale deel van de gastrocnemius daarentegen zal bij het merendeel van de personen eerder hypertrofie vertonen (figuur 9) (17).
De CK-waarde kan hier eveneens verstoord zijn met een gemiddelde waarde van 4500 UI/L. Patiënten kunnen lijden aan een ischemische hartziekte, en daarenboven kan in het vroege stadium van de ziekte soms myoglobinurie aangetoond worden (1,17).
Vraagstelling
Binnen deze masterproef ligt de nadruk voornamelijk op het beschrijven van de myopathologische verschijnselen aan de hand van spierbiopsie analyses, in twee cohorten van patiënten met de types 2A, 2I, 2J en 2L gerekruteerd uit het Universitair Ziekenhuis van Gent. Aan de hand van western blot, immuno- en histochemische technieken zal nagegaan worden hoe de differentiële diagnose van de verscheidene subtypes zich karakteristeert, alsook zal uitgezocht worden welke elementen cruciaal zijn naar de diagnostiek van LGMD toe.
Figuur 9: Atrofie van de benen met hypertrofie ter hoogte van het lateraal deel van de musculus gastrocnemius bij een patiënt met
LGMD2L (17).
Methodologie
1. Literatuuronderzoek
Vooraleer overgegaan wordt tot het onderzoeken van de spierbiopsieën aan de hand van de verscheidene technieken, wordt er een uitgebreid literatuuronderzoek opgestart. Pubmed, Embase en Google Scholar zijn de voornaamste geraadpleegde bronnen alsook heeft het standaardwerk over myologie (Myology: Basis and clinical, third edition, Engel,A. en Franzini-Armstrong,C.) in belangrijke mate bijgedragen tot het bekomen van de nodige achtergrond. Met behulp van de op deze manier verkregen informatie, kan gestart worden met het experimentele luik, met name laboratoriumonderzoek. Voornamelijk wordt gebruik gemaakt van algemene histologische basiskleuringen, en immunofluorescentie. Om dit mogelijk te maken is het noodzakelijk om te beschikken over goede biopten.
2. Spierbiopsie
2.1 Kenmerken van een goede biopsie
De spiergroep die meest geschikt is om te biopseren is een spier met milde tot matige zwakte bij klinisch onderzoek. Dit betekent een rating score van -1 tot -2 op de Mayo Clinical Examination scale (1).
Het is van belang dat het weefsel niet te ernstig beschadigd is, aangezien contractiele elementen dan reeds vervangen kunnen zijn door fibreus- of vetweefsel. Het biopt moet vrij zijn van trauma, alsook mag de te onderzoeken spier een niet te hoge werkload hebben, aangezien dit aanleiding kan geven tot secundaire myopathologische veranderingen in het kader van degeneratieve atrofie.
De procedure gebeurt onder lokale anesthesie met 2% lidocaïne, waarbij de spier zelf niet wordt geïnjecteerd, aangezien dit kan leiden tot secundaire veranderingen. Het te onderzoeken biopt wordt verwijderd met een scalpel, waarbij de afmetingen variëren naargelang het gewenste onderzoek. Voor lichtmicroscopie worden meestal de afmetingen 0,6 op 0,6 cm gehanteerd. Voor genetische onderzoeken, alsook voor blottechnieken bestaan er geen standaardafmetingen, maar over het algemeen worden biopten met een fatsoenlijke dikte gebruikt (1).
2.2 Histochemische kleuringen
Een analyse van een spierbiopsie aan de hand van verscheidene kleuringen, kan pathologische reacties van een spier tevoorschijn brengen onder een lichtmicroscoop. Voor
het spierweefsel maken we gebruik van vriescoupes, aangezien deze doorgaans het nauwkeurigste onderzoek toelaten.
2.2.1 Hematoxyline & eosine
Dit is de meest gebruikte histologische kleuring die gehanteerd wordt binnen de routine pathologie. De basische kleurstof, hematoxyline, kleurt acidofiele structuren in paarsachtig blauw. Dit wil zeggen dat nuclei, ribosomen en ruw endoplasmatisch reticulum een sterke affiniteit hebben hiervoor, omwille van hun hoog gehalte aan RNA, respectievelijk DNA. In contrast staat eosine, een zure stof die aan de basische structuren een roze kleur toekent. De meeste cytoplasmatische eiwitten zijn basisch, vandaar dat we over het algemeen kunnen stellen dat het cytoplasma een roze tint zal verkrijgen bij deze histologische kleuring (18). Dergelijke kleuring zal binnen dit onderzoek voornamelijk gehanteerd worden om fibrose, atrofie en geïnternaliseerde nuclei aan te tonen (18).
2.2.2 PAS
Deze afkorting staat voor periodic acid-schiff reaction, een kleuring die past in het kader van de histochemische technieken, aangezien het specifieke componenten van cellen en weefsels aankleurt. Dergelijke techniek is van onschatbare waarde voor het begrip van de celstructuur en – functie, alsook is het niet weg te denken voor het stellen van een pathologische diagnose op aangetaste weefsels. Oxidatieve werking van perjoodzuur (HIO4) op de 1,2-glycolgroepen in de glucoseresiduen, is de basis van de reactie. De ontstane aldehyde groepen gaan vervolgens reageren met Schiff tot een onoplosbaar complex, waardoor de complexe koolhydraten weergegeven worden als dieprood, traditioneel omschreven als magenta. Het glycogeen, de intracellulaire opslagvorm van koolhydraten in onder andere spiercellen kan eveneens PAS-positief zijn (18).
2.2.3 Gomori trichoom
Deze kleuring is voornamelijk van belang voor het aantonen van bindweefsel, meer bepaald vindt er een binding plaats op collageen, onderdeel van de extracellulaire matrix. Zoals de naam impliceert, kleurt deze methode het weefsel in drie kleuren. Nuclei en andere basofiele structuren worden blauw aangekleurd, collageen groen of blauw afhankelijk van de gebruikte techniek. Als laatste hebben we nog de rode kleur die het cytoplasma, de spieren, erythrocyten en keratine markeert (18).
2.2.4 Mitochondriale kleuringen
Succinaat-dehydrogenase en NADH-dehydrogenase zijn twee enzymen die aanwezig zijn ter hoogte van de mitochondria, en naargelang het type vezel kan er een variatie optreden in aanwezigheid ervan. Aan de hand van kleuringen gericht tegen dergelijke enzymen, kan een overwicht aangetoond worden in type I of II vezels (18).
De werkingswijze van een skeletspier varieert van het ene deel van het lichaam tot het andere. Sommige spiergroepen, zoals deze die betrokken zijn bij de lichaamshouding, moeten bijna continu samentrekken, terwijl anderen, zoals de extra-oculaire spieren, snelle kortstondige bewegingen maken. De meeste spieren bevatten een mengsel van verschillende vezeltypen. Type I, ook wel de aerobe vezel genaamd, bevat een grote hoeveelheid mitochondria, alsook een grote hoeveelheid van myoglobine, een opslagplaats voor zuurstof, analoog aan hemoglobine. In contrast staat daar tegenover het type II, of de anaerobe vezel. Zij bevatten minder mitochondria en myoglobine, maar zijn daarentegen wel rijk aan glycogeen en glycolytische enzymen. Type II heeft een predominantie in spieren die verantwoordelijk zijn voor een intense maar sporadische contractie, zoals de biceps en triceps van de arm (1,18). Is het mogelijk deze voornaamste types van vezels te onderscheiden van elkaar? Hiervoor kan zoals vermeld gebruik gemaakt worden van succinaat-dehydrogenase en NADH-dehydrogenase. Bijkomend kan enerzijds de PAS-kleuring gebruikt worden, maar het kan ook aan de hand van het myosine ATPase. Dit betreft een groep van enzymen die werken als een ionenpomp ter hoogte van het membraan. De kleuring detecteert voornamelijk het ATPase aanwezig in mitochondria, wat een onderscheid tussen de verscheidene types mogelijk maakt (18).
De biopsies worden onderworpen aan ATP-reacties bij een pH van 4,3 4,6 en 9,4 aangezien de pH-labiliteit van deze reactie een differentiatie van de vezels toe laat in vier types. Type I is base labiel en zuur stabiel, terwijl type II net het tegenovergestelde is, met een graad van variatie tussen de verschillende vezels, waardoor nog een onderscheid gemaakt wordt van A tot C (18).
Het typische dambord of mozaïekpatroon van een menselijke spier, dat bekomen wordt tijdens deze kleuring is het resultaat van de symmetrische verdeling van de donkere type I en de lichtere type II vezels. Binnen de differentiële diagnose van LGMD, is het van belang om te kijken of er al dan niet belangrijke predominantie aanwezig is van één welbepaald vezeltype (18).
Cytochrome c oxidase behoort eveneens tot de mitochondriale kleuringen. Het betreft een enzym dat een belangrijke rol speelt binnen de mitochondriale oxidatieve fosforylering, van belang voor de zuurstofwisseling van de cellen (18). Een deficiëntie ervan ter hoogte van de spiercellen kan leiden tot spierzwakte, wat een element kan zijn binnen LGMD (1).
2.3 Immunohistochemische en –fluorescente kleuringen (IHC en IF)
Voor het aankleuren van specifieke celcomponenten kunnen we eveneens gebruik maken van een antilichaam gericht tegen een bepaald antigen. Dit kan bijvoorbeeld een eiwit betreffen dat deficiënt is in de LGMD-patiënt, waardoor als resultaat al dan niet een verminderd signaal aanwezig zal zijn. Het is belangrijk om de coupes, die meestal bewaard worden op een temperatuur van -80°C te fixeren in aceton voor 3 minuten. Daarna wordt PBS (phosphate buffer saline) met het humaan- en ezelserum blokkeringsmedium toegevoegd gedurende 30 minuten.
Het primair antilichaam wordt verdund in het blokkeringsmedium aangebracht. Het antilichaam bindt op het epitoop van het eiwit van interesse. Voor de uiteindelijke visualisatie wordt gebruik gemaakt van een secundair antilichaam, gelabeld met een fluorescerende stof voor IF, dat gericht is tegen het antilichaam van de diersoort waarin het primaire antilichaam werd opgewekt.
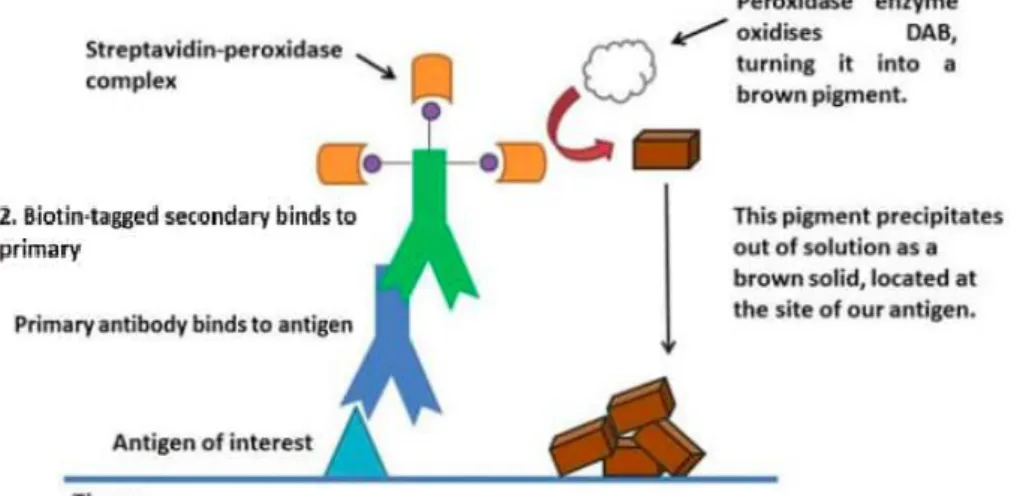
Binnen de masterproef is één IF-kleuring uitgevoerd waarbij twee weefselcoupes van normaal spierweefsel vergeleken worden met de coupes van een patiënt met LGMD2A. Het eerste antilichaam is afkomstig uit een muis en is gericht tegen dystrofine, een eiwit dat zich bevindt ter hoogte van het sarcolemma. Het tweede antilichaam is afkomstig uit een konijn, en kleurt de cellen die NCAM (neural cell adhesion molecule) positief zijn, een regeneratie merker. Bijkomend is er IHC-kleuring uitgevoerd met visualisatie van ANO5, een eiwit dat defect is bij patiënten met LGMD2L. De procedure is dezelfde als hierboven vermeld, maar voor een IHC-kleuring wordt gebruik gemaakt van gebiotinyleerd secundair anti-konijn, anti-muis antilichaammengsel gevolgd door een streptavidin-peroxidase. Tot slot wordt een DAB(3,3′-Diaminobenzidine)-substraat buffer samen met een DAB-chromogeen toegevoegd, waardoor een bruine kleuring verkregen wordt (figuur 106).
2.4 Western blot
Binnen deze masterproef wordt in belangrijke mate gebruik gemaakt van western blots. Het principe hiervan is in hoofdzaak de separatie van eiwitten op basis van hun gewicht of lengte door middel van een gel elektroforese, de transfer naar een nitrocellulose membraan en de selectieve detectie van een antigen. Dit betreft een routine methode voor de analyse van proteïnen, en hangt in belangrijke mate af van de specificiteit van antilichaam-antigen interactie en is een bruikbaar principe voor de kwantitatieve identificatie van specifieke eiwitten en hun moleculaire massa. Omwille van het feit dat het kwantitatieve aspect hier op de voorgrond staat, kunnen er vergelijkingen gemaakt worden tussen stalen, wat binnen deze masterproef voor de differentiële diagnose van LGMD van essentieel belang is.
Alvorens er kan overgegaan worden tot deze techniek is het belangrijk een extract te maken van eiwitten, gehaald uit een cellijn of biopt. Eenmaal dit is gebeurd is het denatureren van de eiwitten een vereiste binnen western blot, wat kan gebeuren op basis van een loading buffer SDS (sodium dodecyl sulfaat) of LDS (lithium dodecyl sulfaat). Dit zorgt voor het ontvouwen van het eiwit en het aanbrengen van een negatieve lading op de eiwitten, waardoor er een aantrekking kan plaatsvinden naar de positieve pool na het aanleggen van een elektrisch veld. Naast de loading buffer, is het van belang de eiwitten gedurende een tweetal minuten te koken. Door de temperatuursverhoging verliest het verder zijn ruimtelijke vorm zodat een lineaire structuur bekomen wordt.
Dit levert een staal op bruikbaar voor elektroforese. Voor de twee western blots (ANO5 en sarcoglycanen) binnen het experimenteel luik wordt een 10% bis tris polyacrylamide gel en Mops buffer gebruikt. De concentratie determineert de resolutie van de gel. Hoe groter de concentratie, des te beter de resolutie is van proteïnen met een laagmoleculair gewicht.
Figuur 10: Grafische voorstelling van een immunohistochemische kleuring, waarbij de verschillende lagen van bindende antilichamen, die het signaal versterken op de plaats van het antigeen van interesse, weergegeven worden. De oxidatie van DAB (3,3′-Diaminobenzidine) veroorzaakt een bruin pigment dat neerslaat op de locatie
De stalen die we wensen te onderzoeken worden geladen in slotjes in de gel. Een of meerdere banen worden normaliter gereserveerd voor een marker, wat een mengsel is van eiwitten met gekend moleculair gewicht die dankzij aankleuring zichtbare banden vormen (SeeBlue™ Plus2 Pre-stained Protein Standard). Zo kan gevolgd worden in hoeverre de eiwitbanden al tot stand zijn gekomen na het aanleggen van een elektrisch veld over de gel. Proteïnen met een kleiner gewicht of lengte zullen sneller migreren naar de positieve pool dan grotere eiwitten. Deze verschillende elektroforetische mobiliteiten scheiden zich af in banden binnen elke rij (figuur 117).
De eiwitten migreren, maar zijn nog niet visueel aantoonbaar. Na blokkering met BSA (bovine serum albumine) of melkpoeder aangelengd met TBS (tris-buffered saline), worden antilichamen gericht tegen een specifiek epitoop van het proteïne toegevoegd. Dit staat bekend als primaire antibody incubatie, waarbij er specifieke commerciële monoclonale antilichamen gebruikt worden, bekomen uit een muis hybridoma (sarcoglycanen test) of geïmmuniseerde geiten of konijnen (ANO5 test). Dit zorgt nog steeds niet voor een visualiseerbaar resultaat. Daarvoor dient een secundair antibody incubatie. Dit houdt in dat het specifiek gebonden antilichaam gevisualiseerd wordt door de blot te behandelen met een tweede antilichaam gericht tegen het eerste. Dit is gelabeld met een welbepaald enzym, waardoor een kleuring tot stand kan komen. Voor de test met betrekking tot ANO5 gebeurt de beeldvorming door middel van ECL, wat staat voor enhanced chemiluminescence, waarbij tijdens een chemische reactie energie vrijkomt onder de vorm van licht (figuur 128). Dit is
visualiseerbaar met een toestel genaamd Chemidoc (Biorad).
7https://www.creative-diagnostics.com/Sample-Gel-Preparation.htm
Tabel 2: Schematische voorstelling van de gehanteerde antilichamen met herkomst en verdunning voor immunofluorescentie en/of western blot.
antilichaam herkomst verdunning
immunofluorescentie verdunning western blot DYSFERLINE Novocastra 7,2 µg/ml 0,9 µg/ml DYSTROFINE 1/2/3 Novocastra 50 µg/ml 10 µg/ml COLLAGEN 6 Millipore 50 µg/ml CALPAÏNE 3 Novocastra 10 µg/ml 10 µg/ml CAVEOLINE 3 BD Biosciences 5 µg/ml 2,5 µg/ml SARCOGLYCANEN A,B,D,G Leica :148 µg/ml; :228 µg/ml :44 µg/ml; :132 µg/ml :18,5 µg/ml; : 28,5 µg/ml; : 18 µg/ml; : 16,5 µg/ml
DYSTROGLYCAN A,B Leica 20 µg/ml 10 µg/ml
ANOCTAMIN 5 LSBio 4,5 µg/ml 0,9 µg/ml MHC-I Dako 9,4µg/ml 0, 47 µg/ml MEMBRANE ATTACK COMPLEX MAC Dako 0,45µg/ml CD4 Dako 1 µg/ml
RABBIT ANTI-NCAM Chemicon 4 µg/ml
CD3 Dako 2,4 µg/ml CD8 Novocastra 20 µg/ml CD1C Santa Cruz Biotechnology 2 µg/ml CD68 SantaCruz Biotech 4µg/ml
Figuur 12: De incubatie van het primair en secundair antilichaam gevolgd door visualisatie met behulp van chemiluminiscentie.
3. Patiënten
Binnen dit onderzoek worden twee cohorten bestudeerd. Eén cohorte van acht patiënten met vier verschillende recessieve types LGMD, zijnde 2A, 2I, 2J en 2L en een tweede cohorte met vijf LGMD2L patiënten is gekenmerkt door een ANO5 mutatie. Iedere patiënt is gerekruteerd in het Universitair Ziekenhuis van Gent. Op het spierweefsel worden verscheidene onderzoeken verricht, waaronder IHC en IF-kleuringen, evenals western blot.
Tabel 3: Schematische voorstelling van de cohorte van acht patiënten met respectievelijk LGMD2A, 2I, 2J en 2L.
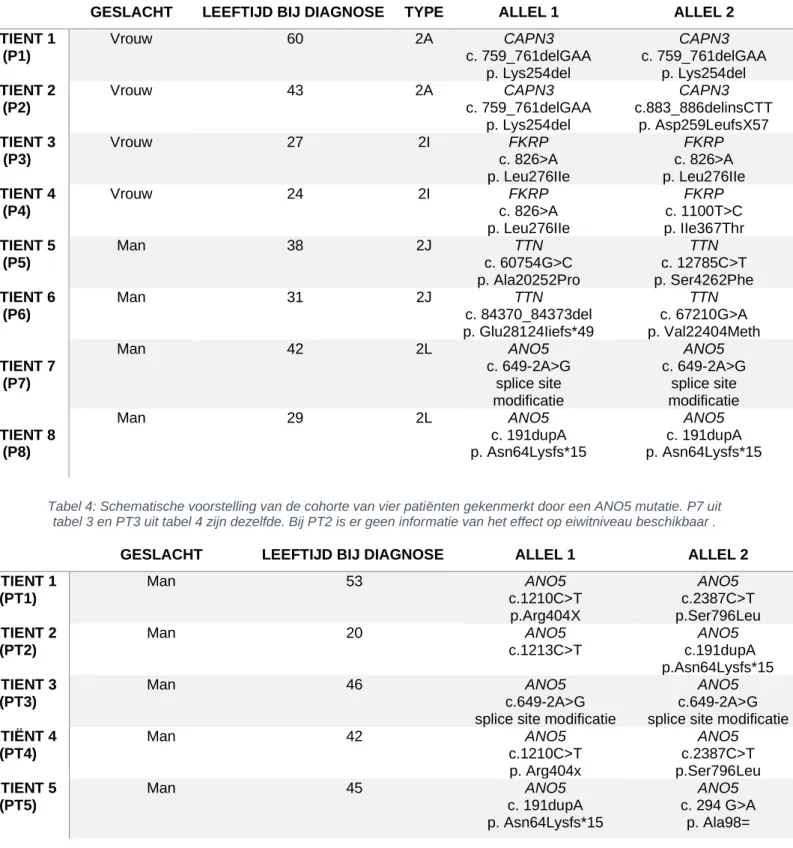
Tabel 4: Schematische voorstelling van de cohorte van vier patiënten gekenmerkt door een ANO5 mutatie. P7 uit tabel 3 en PT3 uit tabel 4 zijn dezelfde. Bij PT2 is er geen informatie van het effect op eiwitniveau beschikbaar .
GESLACHT LEEFTIJD BIJ DIAGNOSE TYPE ALLEL 1 ALLEL 2
PATIENT 1 (P1) Vrouw 60 2A CAPN3 c. 759_761delGAA p. Lys254del CAPN3 c. 759_761delGAA p. Lys254del PATIENT 2 (P2) Vrouw 43 2A CAPN3 c. 759_761delGAA p. Lys254del CAPN3 c.883_886delinsCTT p. Asp259LeufsX57 PATIENT 3 (P3) Vrouw 27 2I FKRP c. 826>A p. Leu276IIe FKRP c. 826>A p. Leu276IIe PATIENT 4 (P4) Vrouw 24 2I FKRP c. 826>A p. Leu276IIe FKRP c. 1100T>C p. IIe367Thr PATIENT 5 (P5) Man 38 2J TTN c. 60754G>C p. Ala20252Pro TTN c. 12785C>T p. Ser4262Phe PATIENT 6 (P6) Man 31 2J TTN c. 84370_84373del p. Glu28124Iiefs*49 TTN c. 67210G>A p. Val22404Meth PATIENT 7 (P7) Man 42 2L ANO5 c. 649-2A>G splice site modificatie ANO5 c. 649-2A>G splice site modificatie PATIENT 8 (P8) Man 29 2L ANO5 c. 191dupA p. Asn64Lysfs*15 ANO5 c. 191dupA p. Asn64Lysfs*15
GESLACHT LEEFTIJD BIJ DIAGNOSE ALLEL 1 ALLEL 2
PATIENT 1 (PT1) Man 53 ANO5 c.1210C>T p.Arg404X ANO5 c.2387C>T p.Ser796Leu PATIENT 2 (PT2) Man 20 ANO5 c.1213C>T ANO5 c.191dupA p.Asn64Lysfs*15 PATIENT 3 (PT3) Man 46 ANO5 c.649-2A>G splice site modificatie
ANO5
c.649-2A>G splice site modificatie PATIËNT 4 (PT4) Man 42 ANO5 c.1210C>T p. Arg404x ANO5 c.2387C>T p.Ser796Leu PATIENT 5 (PT5) Man 45 ANO5 c. 191dupA ANO5 c. 294 G>A
Resultaten
Een vergelijking wordt gemaakt tussen patiënten met hetzelfde LGMD-subtype op basis van klinische, moleculaire, histologische en immunofluorescente karakteristieken. De western blot wordt eveneens besproken. Op pagina 40 is een samenvattende tabel hierover terug te vinden per patiënt (tabel 5).
1. LGMD2A
1.1 Klinische en genetische karakteristieken
Patiënt 1 betreft op het ogenblik van diagnose een 60-jarige vrouw met een CAPN3 homozygote mutatie (c.759_761 delGAA p.lys254del) in exon 5. Dit betekent dat op moleculair niveau er een deletie plaatsvindt van GAA, waardoor het aminozuur lysine op plaats 254 niet meer aanwezig is. Om de klinische significantie hiervan te achterhalen wordt gebruik gemaakt van ClinVar9, een online tool, die toelaat om de relatie na te gaan tussen medisch belangrijke
varianten en fenotypes. Er wordt gekeken welke varianten al dan niet als pathogeen worden beschouwd. Bij deze patiënt is er sprake van een pathogene variant. P1 is de dochter van twee gezonde ouders en ze heeft eveneens één zus en drie broers. Eén broer ondervond zelf reeds symptomen rond de leeftijd van 13 jaar. De eerste klacht bij de patiënt rond de leeftijd van 20 jaar is proximale zwakte ter hoogte van de extremiteiten, alsook rond de schoudergordel. Bijkomend is een duidelijke verhoging merkbaar van het CK-level, waarbij de waarde ongeveer twee maal de maximale waarde van 174 IU/L overtreft. De vrouw bevindt zich momenteel reeds in een rolstoel. Er is eveneens cardiale betrokkenheid, met een recent doorgemaakt ST-elevatie myocardinfarct gecombineerd met ventriculaire fibrillatie. Een drug eluting stent is hiervoor geplaatst, gevolgd door cardiale opvolging op regelmatige basis. Patiënt 2 is een vrouw van 43 jaar op het moment van de diagnose. Ze heeft een CAPN3 heterozygote mutatie (c.759_761delGAA lys254 del en c.883_886delinsCTT p.Asp259Leu) in exon 5 en 6. Er kan gesteld worden dat de eerste mutatie hetzelfde is als P1, maar bijkomend is er sprake van een deletie, met als resultaat dat op nummer 259 het aminozuur aspartaat is vervangen door leucine. Ook hier zijn beide mutaties volgens ClinVar pathogeen. De vrouw in kwestie is de dochter van twee gezonde ouders. Ze komt uit een gezin van vier, waarbij haar broer en twee zussen geen klachten vertonen. Rond de leeftijd van 12 jaar ondervond deze patiënt de eerste klinische tekenen. Het betreft voornamelijk zwakte, gelokaliseerd proximaal aan de bekkengordel met bijkomende klachten ter hoogte van de schouder. Het CK-level (324
IU/L) is ongeveer dubbel zo hoog in vergelijking met de normale waarde. Er is geen cardiale of pulmonale betrokkenheid, maar wel is de vrouw reeds afhankelijk van een rolstoel.
1.2 Histologische karakteristieken
In het kader van de histologie, worden de spierbiopsieën beoordeeld op verschillende elementen en dit met behulp van lichtmicroscopie.
Bij beide patiënten is er een abnormale variatie in de diameter van de spiervezels, waarbij de meeste vezels een eerder atroof aspect vertonen. Bij P1 zijn ze in grotere aantallen aanwezig, terwijl een clustering in kleine groepjes eerder kenmerkend is voor P2. De bindweefselvorming of fibrose zichtbaar aan de hand van de blauwe Gomori trichoomkleuring is bij beide extensief aanwezig. Op pagina 37 is een collage van foto’s terug te vinden betreffende de atrofie en fibrose bij alle patiënten, behalve P2 ontbreekt door het niet voorhanden zijn van weefselcoupes (figuur 27).
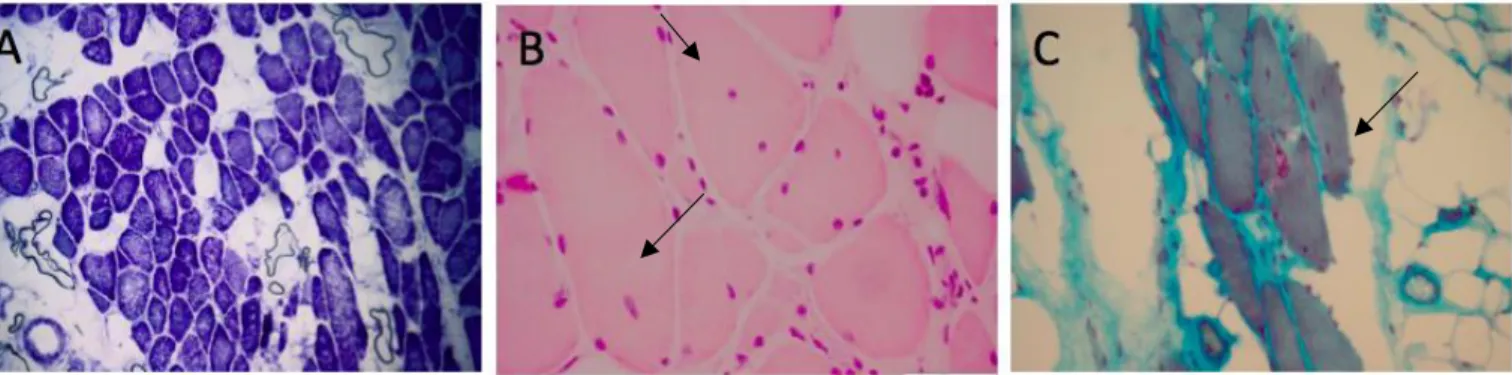
Er is een duidelijke predominantie voor de donker aankleurende type I-vezels bij beide patiënten (figuur 13). Er is geen sprake van COX-deficiëntie. Wat betreft de necrotiserende vezels, kan gesteld worden dat deze niet aanwezig zijn bij P1, terwijl P2 dit in zeldzame mate wel vertoont. Dit weerspiegelt zich via een verhoogde zure fosfatase activiteit in de macrofagen rond de spiervezels (figuur 13). De rimmed vacuoles en interne nuclei (figuur 13) zijn prominenter aanwezig bij P1, terwijl bij P2 abnormale inclusies of vacuolen niet voorkomen of toch in veel mindere mate.
Figuur 13: Histochemische analyse van P1. Foto A: NADH-dehydrogenase kleuring: predominantie donkere type I vezels. Scale bar = 200 m. Foto B: H&E kleuring: interne nucle (pijl)i. Scale bar = 50 m. Foto C: Gomori Trichoom: rimmed vacuole
1.3 Immunofluorescente karakteristieken
MHC-I (major histocompatibility complex I), dat betrokken is in het immuunsysteem, is duidelijk opgereguleerd bij P2. MAC staat voor membrane attack complex en hoort thuis bij het complementsysteem, onderdeel van onze aangeboren immuniteit en zal zich voornamelijk manifesteren ter hoogte van de bloedvaten. Aangezien MHC-I en MAC binnen het kader van inflammatie passen, kan gesteld worden dat er zich inflammatoire kenmerken manifesteren. NCAM (neural cell adhesion molecule) toont regeneratie aan. Deze drie elementen zijn niet gedetermineerd bij P1. MHC-I is sterk positief bij P2, NCAM is daarentegen negatief, terwijl capillairen een verhoogd signaal tonen voor MAC.
Dystrofine is een in het sarcolemma gelegen eiwit waarbij de detectie van het eiwit toevertrouwd wordt aan drie verschillende antilichamen die gericht zijn tegen drie verschillende delen van het eiwit zijnde dys 1, 2 en 3. Bij P1 zien we dat er op IF een verzwakt signaal aanwezig is, maar dit enkel voor dys 1, de overige vertonen een normaal beeld. Bij P2 daarentegen zien we een patroon van mozaïcisme, waarbij bepaalde zones meer signaal capteren dan andere, en dit zowel bij dys 1 als dys 3. Een normaal beeld wordt verkregen voor dys 2.
Vervolgens is -DAG (alfa-dystroglycan), ook wel DAG1 genoemd, zowel bij P1 als bij P2 verzwakt, terwijl de -vorm normaal is (DAG2). SGC (sarcoglycanen) komen voor in vier vormen met name (SGCA), (SGCB), (SGCD) en (SGCG). Bij beide patiënten zijn SGCA SGCB en SGCD normaal. SGCG is niet bepaald. Een zwak signaal wordt verkregen voor dysferline bij P1, terwijl dit normaal is bij P2.
De meeste foto’s kunnen niet getoond worden, aangezien de IF-coupes van beide patiënten niet meer aanwezig zijn binnen de dienst neuropathologie. Daarom wordt een extra IF-kleuring uitgevoerd, waarbij het spierweefsel van P2 vergeleken wordt met dit van een controle persoon (figuur 14). Het gaat over dys 1 in combinatie met NCAM. Het mozaïekbeeld bij dys1 is duidelijk zichtbaar (groene fluorescentie). Bemerk eveneens de split fiber, een gespleten vezel (pijl) wat een kenmerk is binnen LGMD. NCAM, wijzend op regeneratie, is positief bij P2 in de kleinere spiervezels (rode fluorescentie). Bij de controle, zien we over het algemeen een mooie aanwezigheid van dys 1, en geen positief signaal voor NCAM.
1.4 Western blot
Western blots zijn voor P1 niet gebeurd. Bij P2 zijn western blots beschikbaar voor dystrofine, dysferline en CAPN3. De patiënt vertoont een CAPN3 mutatie wat zich vertaalt in een verminderd signaal hiervoor. Eveneens is het signaal verzwakt voor dystrofine 1. Voor dysferline wordt een normaal beeld gevonden.
2. LGMD2I
2.1 Klinische en genetische karakteristieken
P3 met LGMD2I subtype betreft een jonge vrouw van 27 jaar op het ogenblik van de biopsie met een homozygote mutatie ter hoogte van het FKRP gen (c. 826C>A p. Leu276IIe). Dit wil zeggen dat er sprake is van een puntmutatie op niveau 826 waarbij C vervangen wordt door A. Op eiwitniveau zorgt dit ervoor dat het aminozuur leucine vervangen wordt door isoleucine. Deze mutatie is volgens ClinVar pathogeen. Binnen de familiestamboom is er sprake van consanguiniteit en een broer van haar vertoont gelijkaardige symptomen. De patiënte heeft als klinische presentatie proximale zwakte in de onderste ledematen, waarbij links meer dan rechts. Er is atrofie van de spieren van de schoudergordel (trapezius, biceps en triceps), en eveneens scapula alata. Op intern gebied is er momenteel nog geen aantasting van hart en longen.
Figuur 14: Immunofluorescentie analyse van dystrofine 1 (groene fluorescentie) en NCAM (rode fluorescentie) bij P2 en controle. Scale bar = 100 m. Bemerk het mozaïek patroon bij dystrofine 1, wat typerend is voor patiënt 2, alsook
Figuur 15: Foto A: H&E kleuring P3: interne nuclei met een halo (zie pijl). Scale bar = 50m. Foto B: Zure fosfatase kleuring P3: verhoogde activiteit in de macrofagen (roze cellen). Scale bar = 100m.
Foto C: H&E kleuring P4: interne nuclei (zie pijl). Scale bar = 100m.
De tweede persoon (P4) die tot deze categorie behoort is eveneens een jonge vrouw van 24 jaar met een heterozygote mutatie van FKRP (c. 826C>A p.Leu276IIe en c.1100T>C p.IIe367Thr). De eerste mutatie is hetzelfde als bij P3, maar de tweede is een puntmutatie op niveau 1100 waarbij T vervangen wordt door C, met als resultaat dat het aminozuur threonine in de plaats komt van isoleucine. De eerste mutatie is pathogeen, daar waar de tweede onzeker is volgens ClinVar. De familiale voorgeschiedenis is onbekend, ze heeft geen broers of zussen en geen contact meer met haar ouders. Ze heeft Zuid-Afrikaanse roots. De vrouw presenteerde zich voor het eerst op de leeftijd van 15 jaar met klachten van moeilijke gang. Bijkomend was er eveneens zwakte ter hoogte van de proximale spiergroepen en schouders. De patiënte heeft geen cardiale symptomen, maar op pneumologisch gebied is er wel lichte dyspnoe met een FVC (forced vital capacity) van 71%.
2.2 Histologische karakteristieken
Er is een abnormale variatie in spiervezeldiameter. Wat betreft de atrofie kan gesteld worden dat dit eerder zeldzaam is, met een lichte overmaat voor P4. Fibrose is bij beide patiënten niet aanwezig (figuur 27).
Bij P3 zijn er duidelijk necrotiserende vezels aanwezig, maar dit is vooral te visualiseren met behulp van een fosfatase kleuring, die een verhoogde zure fosfatase activiteit aantoont in de macrofagen (figuur 15). Wanneer dit vergeleken wordt met P4, is er bij de laatstgenoemde maar heel weinig necrose. Bij beide patiënten is er geen sprake van rimmed vacuolen of COX-deficiëntie. Daarenboven blijft het dambordpatroon mooi bewaard, aangezien er geen predominantie aanwezig is.
Typerend voor deze patiënten is een opvallende toename van het aantal vezels met interne nuclei. Sommige, vooral bij P3, zijn door een leeg halo (pijl) omringd, maar dwarse sneden hiervan zijn zeldzaam (figuur 15).
2.3 Immunofluorescente karakteristieken
Dys 1, 2 en 3 zijn normaal bij beide patiënten. -DAG (DAG1) daarentegen vertoont bij P3 een mozaïek patroon, terwijl dit bij P4 opgemerkt wordt in een beperkt aantal vezels. In tegenstelling tot -DAG is de -vorm (DAG2) niet afwijkend bij beide personen.
SCG vertoont geen noemenswaardige veranderingen, waardoor kan gesteld worden dat dit normaal is bij beide personen op IF. Dysferline is uitsluitend bij P3 bepaald, maar ook hier zijn er geen afwijkingen aanwezig.
Wat betreft MHC-I zien we dat P3 een positiviteit vertoont voor een aantal vezels, alsook kleuren de arteriolen voor MAC, beide elementen die in de richtingen wijzen van inflammatie. Bij P4 is MHC-I slechts positief bij een zeldzaam aantal vezels. MAC daarentegen vertoont aankleuring ter hoogte van de capillairen (figuur 16).
Wordt er gekeken naar NCAM, is een groot verschil op te merken, aangezien bij P3 heel veel kleine vezels aankleuren, daar waar P4 negatief is (figuur 16).
Figuur 16: Immunofluorescentie analyse van MHC-I, NCAM en MAC bij P3 en P4. . NCAM (rood): P3 kleinere vezels positief, P4 negatief. MHC-I (groen): negatief bij P3 en P4. MAC (rood): arteriolen en capillairen positief bij respectievelijk P3 en P4. Scale bar = 100 m.
2.4 Western blot
Over de western blots zijn van P3 geen gegevens beschikbaar. Wat betreft P4, kan gesteld worden dat dys1 normaal is, alsook zijn er geen afwijkingen bij dysferline en CAPN3.
Binnen het experimenteel luik van de masterproef werd een western blot uitgevoerd voor SGC- (SGCA), - (SGCB) en - (SGCG). Hierbij werd het spierweefsel van P4 vergeleken met een controle (C). Als we kijken naar SCGA ( figuur 18 afbeelding A) zien we een eiwitband die zich donkerder presenteert rond de 50 kDA. Dat dit zoveel kDA is kan gezien worden in de figuur naast de western blot (figuur 17), waarbij de bandjes van de gebruikte marker (SeeBlue™ Plus2 Pre-stained Protein Standard) overeenkomen met een welbepaald moleculair gewicht, rekening houdend met de gebruikte gel tijdens de procedure (10% bis-tris MOPS). Er is in vergelijking met de controle geen verminderd signaal aanwezig bij patiënt 4, waardoor er kan gesteld worden dat de western blot van SCGA normaal is.
Bij SGCG ( figuur 18 afbeelding B) wordt een bandje gezien ter hoogte van 97 kDa ongeveer, die bij P4 een verminderd signaal vertoont in vergelijking met de controle. SGCG heeft een verwacht moleculair gewicht van om en bij de 35 kDa, maar daar wordt er geen duidelijk signaal waargenomen. Het hoge gewicht van de band kan verklaard worden door het feit dat de condities mild denaturerend zijn. De interacties tussen de beta, gamma en delta vormen van sarcoglycan zijn vrij sterk, en hoogstwaarschijnlijk detecteert de western blot dit gehele complex. Alfa sarcoglycan (50 kDa) zit losser in het complex, waardoor dit niet mee interageert. Individueel zijn die drie eiwitten 43 (SGCB), en 35 (SGCG en SGCD) kDa, dus dat komt ongeveer overeen met het gewicht dat op de blot gevonden wordt. Zeker aan de uiteinden van een blot is het gewicht niet lineair te bepalen met de merker, dus 97 en 113 is dan binnen de schommeling van het schijnbaar gewicht dat bekomen wordt. De resultaten van de blot blijven eveneens geldig, aangezien het antilichaam de gamma vorm binnen dit sarcoglycan complex detecteert. SGCG is dus wel degelijk verlaagd bij P4, wanneer dit vergeleken wordt met onze controle.
Tot slot werd ook SGCD (figuur 18 afbeelding C) getest en vergeleken tussen P4 en de controle. Een bandje kan gedetecteerd worden rond de 30 kDa, wat overeenkomt met het gekend moleculair gewicht van SGCD. Hier is geen verschil in signaal aanwezig tussen beide personen, waardoor de western blot voor dit eiwit normaal is.
3. LGMD2J
3.1 Klinische en genetische karakteristieken
Patiënt 5 (P5) is een man van 38 jaar op het moment van de biopsie en vertoont een heterozygote TTN mutatie (c. 60754G>C p. Ala20252Pro en c. 12785C>T p. Ser4262Phe). De eerstgenoemde betekent een puntmutatie waarbij G vervangen is door een C op plaats 60754. Dit zorgt voor een verandering in aminozuur van alanine naar proline. Deze verandering wordt door ClinVar als onzeker beschouwd. Dit is ook zo voor de tweede mutatie waarbij op moleculair niveau C vervangen is door T, met als resultaat dat phenylalanine in de plaats komt van serine ter hoogte van 4262. Klinisch presenteert de patiënt zich met gedaalde inspanningstolerantie, vermoeidheid ter hoogte van de spieren en een CK-waarde van 262 IU/l. Hij heeft 1 broer zonder symptomen en geen kinderen.
P6 is een jongeman van 34 jaar met een TTN heterozygote mutatie (c. 84370_84373del p.Glu228124Iiefs en c.67210G>A p.Val22404Meth). De eerste mutatie betekent een deletie van element 84371 en 84372, waardoor een framing shift plaatsvindt, met als gevolg dat het eiwit, het product van dit gen, na dergelijke mutatie geheel veranderd zal zijn. Ondanks deze grote verandering, geeft ClinVar aan dat dit waarschijnlijk pathogeen is, maar niet met 100% zekerheid kan gesteld worden. De tweede fout situeert zich genetisch op niveau 67210 waarbij G vervangen wordt door een A met als gevolg dat op het niveau van het eiwit valine vervangen
Figuur 18:: Western blot van P4 en controle voor SGCA, SGCG en SGCD A = SGCA (50 kDa) en C = SGCD (35 kDa): normaal bij controle en P4. B = SGCG (35 kDa): verminderd signaal bij P4 en normaal bij controle. Figuur 17: Weergave van de moleculaire gewichten per
gebruikte gel bij de marker SeeBlue™ Plus2 Pre-stained Protein.
Figuur 19: Histochemische analyse van cytochroom c-oxidase (COX) en ATPase Ph 4,6 bij P5 en P6 Cox: verminderde aankleuring van vezels bij P5 (pijl), normaal bij P6.
ATPase Ph 4,6: dambord patroon bij P5, predominantie voor type I (donker) bij P6. Scale bar foto A, C en D = 100 m, scale bar foto B = 200 m.
wordt door methionine als aminozuur, wat kan gezien worden als een onzekere variant. Hij is de jongste uit een gezin van 4 en binnen de familie is er geen sprake van consanguiniteit. De man heeft zelf een 5-jarig zoontje, die geen kenmerken vertoont wijzend in de richting van een musculaire aandoening. De patiënt presenteerde zich voor het eerst met een dropvoet rechts op de leeftijd van 24 jaar, waarna verder genetisch onderzoek in de richting wees van LGMD2J, bevestigd door een spierbiopsie. Wat betreft de verdere klinische presentatie is er geen cardiale of pulmonaire betrokkenheid gekend. De CK-waarde vertoont een significante verhoging (454 IU/L).
3.2 Histologische karakteristieken
De atrofie kan enkel in heel lichte mate aangetoond worden bij P5. Er is bijzonder veel fibrose aanwezig bij P6, wat in schril contrast staat met P5. De trichoom kleuring vertoont enkel bij P6 een overmaat aan collageneus bindweefsel. Bij beide zijn er geen regenererende vezels zichtbaar, alsook geen rimmed vacuolen. De COX-deficiëntie (figuur 19) kan wel in lichte mate aangetoond worden, maar dit uitsluitend bij P5. Dit is zichtbaar door het minder aankleuren van bepaalde vezels. Het dambordpatroon wat betreft de verdeling van type I en II vezels is bijzonder goed zichtbaar bij P5, waardoor kan gesteld worden dat er geen predominantie aanwezig is. Bij P6 zijn de grotere vezels eerder type I, waardoor een donkere aankleuring zichtbaar is (figuur 19). Tot slot is er geen necrose merkbaar, en interne nuclei zijn over het algemeen maar schaars aanwezig.
3.3 Immunofluorescente karakteristieken
Immunofluorescente dystrofine kleuringen zijn normaal voor P5, terwijl een zwak dys1 en dys3 signaal verkregen wordt bij P6. DAG1, alsook SGCA, SGCB, SGCG en SGCG zijn normaal bij beide patiënten. MHC-I en MAC zijn bij beide personen negatief. NCAM kleuring is verschillend: P5 is negatief terwijl P6 duidelijke positieve kleine vezels vertoont. Figuur 20 en 21 visualiseren de voornaamste verschillen, alsook gelijkenissen tussen beide patiënten.
Figuur 20: Immunofluorescente analyse van NCAM, DAG1 en MAC bij P5 en P6. Scale bar = 100 m NCAM (rood): negatief bij P5, positieve vezels bij P6.
DAG1 (groen): normaal bij P5 en P6. Bemerk het lager signaal bij P5, wellicht omwille van technische redenen. MAC (rood): negatief bij P5, positief ter hoogte van de capillairen bij P6.