Europese Farmacopee
Gouden standaard bij de bereiding van geneesmiddelen
Rapport 370020001/2009M.E. Kubbinga | P.M.J.M. Jongen | E.K. de Rooij-Lamme | D. de Kaste
RIVM Rijksinstituut voor Volksgezondheid en Milieu Postbus 1 3720 BA Bilthoven www.rivm.nl
RIVM rapport 370020001/2009
Europese Farmacopee
Gouden standaard bij de bereiding van geneesmiddelen
M.E. Kubbinga P.M.J.M. Jongen E.K. de Rooij-Lamme D. de Kaste Contact: D. de Kaste
Centrum voor Kwaliteit van Chemisch-Farmaceutische Producten Dries.de.Kaste@rivm.nl
Dit onderzoek werd verricht in opdracht van de Inspectie voor de Gezondheidszorg (IGZ), in het kader van het project V/370020: Europese Farmacopee
© RIVM 2009
Delen uit deze publicatie mogen worden overgenomen op voorwaarde van bronvermelding: 'Rijksinstituut voor Volksgezondheid en Milieu (RIVM), de titel van de publicatie en het jaar van uitgave'.
Rapport in het kort
Europese Farmacopee
Gouden standaard bij de bereiding van geneesmiddelen
Geneesmiddelen die in Europa gemaakt en gebruikt worden, moeten voldoen aan de wettelijke eisen van de Europese Farmacopee. Als een dergelijk wetboek zou ontbreken, zou de regelgeving over de toelating en productie van geneesmiddelen onduidelijk worden, met mogelijke risico’s voor de volksgezondheid als gevolg. Het wettelijke karakter zorgt er namelijk voor dat de kwaliteit van geneesmiddelen in al deze landen gelijkwaardig en uitwisselbaar is. De Europese Farmacopee maakt het hierdoor makkelijker dat goede geneesmiddelen voor mens en dier beschikbaar zijn. Dit blijkt uit onderzoek van het RIVM, dat in opdracht van Inspectie voor de Gezondheidszorg (IGZ) het belang van de wettelijke standaard heeft beschreven.
Voor het onderzoek zijn Nederlandse experts ondervraagd die een bijdrage leveren aan de Europese Farmacopee. Zij zijn afkomstig van onder meer de (semi-)overheid en de farmaceutische industrie. Zij vinden het belangrijk dat Nederland actief bijdraagt aan het ontwikkelen en in stand houden van de Europese Farmacopee. Op deze manier laat Nederland zijn stem horen en kan het ervaringen met andere geneesmiddelenproducerende landen uitwisselen. Ook vinden de experts het van grote waarde dat de Europese Farmacopee als openbare regelgeving om geneesmiddelen te bereiden, blijft bestaan.
Abstract
European Pharmacopoeia
Golden standard for the preparation of medicines
All medicines produced and used in Europe must comply with the legal requirements imposed by the European Pharmacopoeia. If such a law book would not exist, then the legal requirements for the marketing authorisation and the manufacture of medicines would become unclear, with possible risks to public health. The legal nature of these requirements ensures that medicines are of comparable quality and interchangeable throughout Europe. The European Pharmacopoeia consequently facilitates the accessibility of high quality medicines to Europeans and their animals. This is a conclusion drawn from an RIVM study, carried out on behalf of the Dutch Health Care Inspectorate (IGZ), which describes the importance of this legal standard.
Dutch experts who contribute to the European Pharmacopoeia were interviewed for this study. These experts were mainly representatives from (semi-)governmental organisations and the pharmaceutical industry. They consider it important that the Netherlands actively contributes to the development and revision of the European Pharmacopoeia. In this way the Netherlands can voice its opinion and exchange experiences with other medicine-producing countries. The experts consider it extremely important that the European Pharmacopoeia continues to be a public document which regulates the requirements of medicines.
Inhoud
Samenvatting 7 Summary 9 Afkortingen 11 1 Inleiding 13 1.1 Aanleiding en doelstelling 13 1.2 Werkwijze 132 Kaders rond de Europese Farmacopee 15
2.1 Historisch kader 15
2.1.1 Behoefte aan standaardisatie 15
2.1.2 Verdrag inzake de samenstelling van een Europese Farmacopee 17
2.2 Huidig wettelijk kader 18
2.2.1 Europa 18
2.2.2 De Europese Farmacopee in mondiaal verband 19
2.2.3 Nationale Farmacopees in Europa 20
2.2.4 Overige, internationale farmacopees 20
2.2.5 Internationale harmonisatie 21
2.2.6 De inhoud van Europese Farmacopee in 2008 22
2.3 Huidig organisatorisch kader 23
2.3.1 Raad van Europa 23
2.3.2 Europese Farmacopee Commissie 26
2.3.3 EDQM and HealthCare 28
2.3.4 Europese Unie 30
2.3.5 Ontwikkeling, registratie, keuring en controle van geneesmiddelen 31
3 Nederlandse bijdrage aan de Europese Farmacopee 33
3.1 Wettelijk kader in Nederland 33
3.2 Organisatorisch kader in Nederland 35
3.2.1 Betrokken ministeries 35
3.2.2 Nationale delegatie bij de Europese Farmacopee Commissie 35
3.2.3 Inbreng Nederland 36
3.3 Interviews met belanghebbenden 39
3.3.1 Het werken met de Europese Farmacopee 39
3.3.2 De Europese Farmacopee in relatie tot andere farmacopees 42 3.3.3 De Europese Farmacopee in relatie tot de volksgezondheid 43
3.3.4 De bijdrage van Nederland aan de Europese Farmacopee 45
4 Conclusies 49
4.1 Belang van de Europese Farmacopee 49
Dankbetuiging 50 Literatuur 51 Bijlage 1 Geraadpleegde personen 53 Bijlage 2 Morfine: historische ontwikkelingen in de monografie 56 Bijlage 3 Morfine: huidige monografie 61 Bijlage 4 Glycerine: de Ph. Eur. en de volksgezondheid 65 Bijlage 5 Cijfers betreffende de bijdragen van de diverse lidstaten 71 Bijlage 6 Aanbevelingen voor verbetering 74 Bijlage 7 De ‘ijk-el’ van Goslar 79 Bijlage 8 Afscheidsrede van Van Noordwijk 80
Samenvatting
Geneesmiddelen die in Europa worden gemaakt en gebruikt, moeten voldoen aan de wettelijke eisen van de Europese Farmacopee. Nederland heeft met de ondertekening van het Verdrag inzake het samenstellen van een Europese Farmacopee1 toegezegd een bijdrage te leveren aan deze wettelijke
standaard. In opdracht van de Inspectie voor de Gezondheidszorg (IGZ) heeft het RIVM, Centrum voor Kwaliteit van Chemisch-Farmaceutische Producten (KCF) nu een studie verricht naar het belang van de Europese Farmacopee voor de Volksgezondheid vanuit Nederlands perspectief. Wat zou de schade zijn wanneer de farmacopee als kwaliteitsstandaard zou ontbreken? En is het niet voldoende om de
kwaliteit van de individuele producten bij registratie vast te laten leggen in een registratiedossier? Daarnaast is gevraagd om een uitspraak te doen over de aard en omvang van de Nederlandse inbreng bij het ontwikkelen en in stand houden van de Europese Farmacopee.
In dit rapport wordt allereerst het historisch, wettelijk en organisatorisch kader van de Europese Farmacopee in beeld gebracht. Vervolgens wordt nader ingegaan op de situatie in Nederland en de bijdrage van Nederland aan de Europese Farmacopee. Hiertoe zijn betrokken Nederlandse experts, afkomstig van onder meer de (semi-)overheid en de farmaceutische industrie, geïnterviewd. De Europese Farmacopee is van oudsher gerelateerd aan de volksgezondheid:
• De Europese Farmacopee maakt het mogelijk de eenduidige samenstelling en constante kwaliteit van geneesmiddelen te waarborgen.
• De Europese Farmacopee faciliteert de beschikbaarheid van geneesmiddelen omdat zij toegang biedt tot openbare standaarden die gebruikt kunnen worden in elk stadium van de levensloop van een geneesmiddel; van de ontwikkeling, bereiding, registratie, vrijgifte tot de kwaliteitscontrole. • Uitwisselbaarheid van kwalitatief goede producten wordt mogelijk gemaakt door het feit dat de
Europese Farmacopee geldt voor alle in Europa geproduceerde en gebruikte geneesmiddelen. • De openbaar toegankelijke en in heel Europa geldende Europese Farmacopee overstijgt het
individuele en confidentiële registratiedossier en kan daarom een arbitragefunctie bieden bij geschillen en onduidelijkheden.
• De Europese Farmacopee biedt ten slotte mogelijkheid tot handhaving van Europees aanvaarde kwaliteitseisen door middel van inspecties.
De Europese Farmacopee wordt door de experts gezien als de ‘gouden’ standaard voor de kwaliteit van geneesmiddelen voor mens en dier. De farmacopee is niet statisch, maar wordt aangepast op basis van nieuwe technische ontwikkelingen en inzichten. De Nederlandse experts geven aan dat als een dergelijk wetboek zou ontbreken, de regelgeving over de toelating en productie van geneesmiddelen in Europa onduidelijk zou worden, met mogelijke risico’s voor de volksgezondheid als gevolg. Zij achten een situatie zonder Europese Farmacopee ondenkbaar.
De geïnterviewde experts vinden het belangrijk dat Nederland actief bijdraagt aan het ontwikkelen en in stand houden van de Europese Farmacopee. Zij zijn van mening dat Nederland zijn stem moet laten horen en gebruik moet maken van de kans om ervaringen met andere geneesmiddelenproducerende landen uit te wisselen. De experts denken over het algemeen positief over de werkwijze waarop de Europese Farmacopee wordt samengesteld en onderhouden. Het rapport bevat ten slotte een overzicht van de door deze Nederlandse experts genoemde aanbevelingen voor de toekomst.
1 Nederland is één van de 36 landen van de Raad van Europa die het verdrag van de Raad van Europa: “Het
Summary
All medicines produced and used in Europe must comply with the legal requirements imposed by the European Pharmacopoeia. The Netherlands has pledged to contribute to the creation and maintenance of the European Pharmacopoeia through the signing of a Convention. The RIVM’s Centre for the Quality of Chemical-Pharmaceutical Products (KCF) has been commissioned by the Dutch Health Care Inspectorate (IGZ) to carry out a study on the importance of the European Pharmacopoeia for public health from a Dutch perspective. What would happen if the Pharmacopoeia no longer existed as quality standard? Is it not enough to simply register the quality of individual products in a registration file? In addition to these questions, a statement was requested with respect to the nature and the scope of the Dutch contribution to the development and continuation of the European Pharmacopoeia.
This report starts by describing the historical, legal and organizational framework of the European Pharmacopoeia and goes on to explain the situation in the Netherlands. The working experiences of Dutch professionals within this organizational framework have also been investigated by conducting interviews with the Dutch experts from the European Pharmacopoeia Commission.
The European Pharmacopoeia is associated with public health in various ways:
• The European Pharmacopoeia ensures that the unambiguous composition and continued reliable quality of medicines is guaranteed;
• The European Pharmacopoeia facilitates the availability of medicines by providing access to official standards that can be used at every stage of the lifecycle of a medicine; from the development, preparation, registration, licensing to quality control;
• The European Pharmacopoeia facilitates the interchangeability of high quality medicines (made possible because the rules of the European Pharmacopoeia apply to all medicines used in Europe) • As the European Pharmacopoeia is publicly accessible and applicable throughout Europe, it is able
to extend its activities beyond individual and confidential marketing application dossiers and assume the role of arbitrator in solving conflicts and clarifying misunderstandings.
• Finally, the European Pharmacopoeia has the authority to enforce the accepted European quality requirements through inspections.
The experts regard the European Pharmacopoeia as the public ‘golden’ standard for the quality of medicines for humans and animals in Europe. The European Pharmacopoeia is continually moving forward, making the necessary adjustments to incorporate new developments and insights. If such a law book should cease to exist, then the laws for the admission and production of medicines would become unclear, with possible risks to public health as an outcome. A situation without European
Pharmacopoeia is inconceivable to them.
The Dutch experts interviewed therefore believe that the Netherlands should assume its responsibilities by actively contributing to the continuation of the European Pharmacopoeia. In this way the
Netherlands can voice its opinion and exchange experiences with other medicine-producing countries. The experts are generally positive with respect to the composition and upkeep of the European law book. This report also includes an overview of recommendations for the future put forward by these Dutch experts.
Afkortingen
ANM Adaptation National Monographs
procedure om nationale monografieën over te nemen in de Europese Farmacopee
biologicals biologische geneesmiddelen
geneesmiddelen waarvan de werkzaamheid berust op een biologische grondstof, in tegenstelling tot chemische producten
BD Bureau Diergeneesmiddelen
BP British Pharmacopoeia
Britse Farmacopee
CBG College ter Beoordeling van Geneesmiddelen
Nederlandse registratieautoriteit voor humane en veterinaire geneesmiddelen
CIDC Centraal Instituut voor DierziekteControle-Lelystad
sinds 1 januari 2008 gefuseerd tot Centraal Veterinair Instituut van Wageningen UR
CRS Chemical Reference Substance
CVI Centraal Veterinair Instituut van Wageningen UR
EDQM European Directorate for the Quality of Medicines & HealthCare onderdeel van de Raad van Europa
EMEA European Medicines Agency
Europese registratieautoriteit
EPC Europese Farmacopee Commissie
EU Europese Unie
FDA Food and Drug Administration
Amerikaanse registratieautoriteit
FNA Formularium der Nederlandse Apothekers
GMP Good Manufacturing Practice
ICH International Conference on Harmonisation of Technical
Requirements for Registration of Pharmaceuticals for Human Use samenwerkingsverband van registratieautoriteiten, inspectie en industrie uit Europa, Verenigde Staten en Japan op het gebied van humane geneesmiddelen
IGZ Inspectie voor de Gezondheidszorg
IntPh International Pharmacopoeia
internationale farmacopee uitgegeven door de WHO
JP Japanese Pharmacopoeia, Japanse Farmacopee
KNMP Koninklijke Nederlandse Maatschappij ter bevordering der Pharmacie
de beroepsorganisatie van apothekers in Nederland
LNV Ministerie van Landbouw, Natuur en Voedselveiligheid
MAT monocyten activatie test
specifiek genoemde testmethode, beschreven in de Ph. Eur.
NFA Nationale Farmacopee Autoriteit
PDG Pharmacopoeial Discussion Group
Ph. Eur. Gebruikelijke afkorting van Pharmacopée Européenne Europese Farmacopee
REFPA Regeling eisen farmaceutische preparaten apotheken
RIVM Rijksinstituut voor Volksgezondheid en Milieu
RvE Raad van Europa (Council of Europe)
SPC Summary of Product Characteristics
wetenschappelijke bijsluiter van een geneesmiddel
USP United States Pharmacopeia
Amerikaanse Farmacopee
VICH International Cooperation on Harmonisation of Technical Requirements for Registration of Veterinary Medicinal Products samenwerkingsverband van registratieautoriteiten, inspectie en industrie uit Europa, Verenigde staten en Japan op het gebied van veterinaire geneesmiddelen
VWS ministerie van Volksgezondheid, Welzijn en Sport
WBGB werkgroep voor Biologische Geneesmiddelen en Biofarmaceutica van de NFA
WHO World Health Organisation
wereldgezondheidsorganisatie
1
Inleiding
1.1
Aanleiding en doelstelling
In opdracht van de Inspectie voor de Gezondheidszorg heeft het RIVM, Centrum voor Kwaliteit van Chemisch-Farmaceutische Producten (KCF) een studie verricht naar het volksgezondheidsbelang van de Europese Farmacopee. Wat zou de schade zijn wanneer de farmacopee als kwaliteitsstandaard zou ontbreken? En is het niet voldoende om de kwaliteit van de individuele producten bij registratie vast te laten leggen in een registratiedossier? Daarnaast is gevraagd om een uitspraak te doen over de aard en omvang van de Nederlandse inbreng bij het ontwikkelen en in stand houden van de Europese
Farmacopee.
1.2
Werkwijze
Allereerst wordt in dit rapport achtergrondinformatie gegeven over de historie en het wettelijk kader van de Europese Farmacopee (Ph. Eur.). Daarnaast is een overzicht gemaakt van de betrokken organisaties rond de Europese Farmacopee.
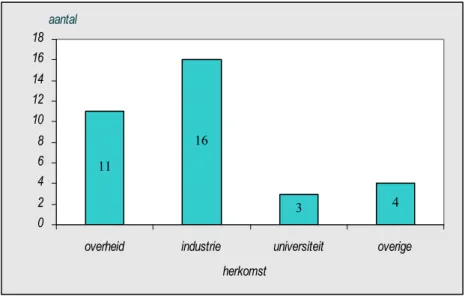
De rol van Nederland en de Nederlandse inzet bij het onderhouden van de Europese Farmacopee krijgt aandacht in hoofdstuk 3. Om inzichtelijk te maken wat voor direct betrokkenen in Nederland het belang is van het bestaan van de Europese Farmacopee zijn interviews gehouden. Nederlandse experts en specialisten uit de expertgroepen van de Europese Farmacopee Commissie en andere belanghebbenden afkomstig uit de farmaceutische industrie en overheid zijn hiervoor benaderd. Zie Bijlage 1 voor een overzicht van geraadpleegde instanties en personen.
Disclaimer
De informatie in hoofdstuk 3 is gebaseerd op gesprekken met diverse personen genoemd in Bijlage 1. De verantwoordelijkheid voor de interpretatie van de interviews en gerelateerde conclusies ligt echter bij de auteurs van dit rapport. De beschreven meningen vormen niet noodzakelijkerwijs de letterlijke weergave van het interview. Daar waar een geïnterviewde wel letterlijk geciteerd is, is dit aangegeven met aanhalingstekens.
2
Kaders rond de Europese Farmacopee
2.1
Historisch kader
Het woord farmacopee is afgeleid van het Griekse woord pharmakopoios waarin de woorden voor geneesmiddel, pharmakon, en maken, poe-ein, terug te vinden zijn. Volgens het Van Dale Groot woordenboek is een farmacopee een ‘officieel, van staatswege uitgegeven handboek met voorschriften voor de bereiding van geneesmiddelen en de vereisten waaraan zij moeten voldoen’. De eerste
vermelding van het woord farmacopee in de Van Dale uit 1864 zag eruit zoals in Figuur 2.1 is weergegeven.(1)
† Pharmaceut, m. (-en), artsenijbereider, -kenner. *...CEUTISCH, bn. artsenijkundig. *...CIE, v. kennis der geneesmiddelen en van hunne bereidingen. *...CON, o. geneesmiddel; tooverdrank; vergif. *...COPOEA, v. handboek der artsenijbereiding; wat de artsenijkunde omvat; - pauperum,
voorschriften betreffende de armen-apotheken (zoo als de berekening van de prijzen der geneesmiddelen, enz.)
Figuur 2.1 De eerste definitie van het woord pharmacopoea in de Van Dale uit 1864 (1)
2.1.1
Behoefte aan standaardisatie
“Alsoo myne Heeren van den Gerechte bericht worden van de groote ongeregeltheid, die er by de Apothekers is in 't prepareren der Medicamenten, daer uyt ontstaende, dat men geen generael voorschrift heeft, waer naer ider winckel gehouden is, syne Geneesdrancken te bereyden: en dat het door de groote der stad en de mennichte der winckelen den Doctoren tot noch toe geensints mogelyck is geweest te weten, wat in elcke winckel was, veel min, hoe elck Medicament aldaer bereyd werde. Door welcke onkunde de Genees-konst in merckelicke onsekerheid geraeckt is, ende de Siecken mits dien in groot gevaer souden hebben konnen vervallen, tot grooten ondienst van de Gemeente, ende geen kleyne dis reputatie der Doctore n...”
Figuur 2.2 Aankondiging van de op 29 april 1636 uitgevaardigde Amsterdamse Farmacopee (2-3)
Van oudsher streeft men bij het voorschrijven en bereiden van geneesmiddelen naar standaardisatie van preparaten. Het niveau waarop deze standaardisatie plaatsvond en plaatsvindt, hangt samen met de politieke ontwikkelingen. De eerste farmacopee ter wereld zou in 1498 of 1499 verschenen zijn in Florence onder de naam ‘Nuovo receptario’ (het nieuwe receptenboek). Allereerst in het Italiaans, later ook in het Latijn, zodat het voor alle beroepsbeoefenaren die het Latijn beheersten toegankelijk werd.(4)
De historie van de farmacopee in Nederland begon in de 17e eeuw. Ten tijde van de opkomst van de grote steden ontstonden samenwerkingsverbanden van artsen en apothekers. Deze beroepsbeoefenaren gingen hun voorschriften standaardiseren en vastleggen in de stadsfarmacopee. Hieruit is bijvoorbeeld in 1636 de Amsterdamse Farmacopee voortgekomen. Uit de aankondiging van de Amsterdamse Farmacopee door het stadbestuur (zie Figuur 2.2) blijkt dat er ook toen al grote behoefte was aan transparante informatie en aan geharmoniseerde, gestandaardiseerde voorschriften. Dit om te zorgen
dat elke patiënt overal dezelfde kwaliteit medicijnen kreeg.(2-3) Ook steden als Utrecht, Haarlem, Den Haag en Leiden ontwikkelden een naslagwerk dat gebruikt werd door voorschrijvers (artsen) en bereiders (apothekers). In diezelfde periode werden bijvoorbeeld ook lengtematen meer
gestandaardiseerd. Zo werd op het marktplein van Goslar (Duitsland) de ‘el’ als lengtemaat openbaar en voor iedereen op de markt geldend gehangen (zie Bijlage 7).
Figuur 2.3 Afbeeldingen van de eerste Amsterdamse Farmacopee; links een afbeelding in het boek, rechts de titelpagina (2)
De behoefte aan standaardisatie kwam voort uit een combinatie van handels- en veiligheidsmotieven. Enerzijds wilde men de betrouwbaarheid van de preparaten qua werkzaamheid en veiligheid bewaken. Dit deed men door het vastleggen van samenstelling en bereidingsvoorschriften. Anderzijds wilde men voorkomen dat te strenge eisen tot monopolyposities van bepaalde bereiders zouden leiden. De vastgelegde standaarden vermeldden daarom minimale kwaliteitseisen voor producten en grondstoffen en gingen niet tot de uiterst haalbare kwaliteit.
In de 19e eeuw werden de stadsfarmacopees samengevoegd, geharmoniseerd en ontstond een nationale farmacopee. In eerste instantie was dit de Bataafse Farmacopee (1805). Vervolgens werd in 1851 de eerste Nederlandse Farmacopee (Ph. Ned.) uitgegeven. Een vergelijkbare ontwikkeling leidde ook in andere Europese landen tot de samenstelling van nationale formularia en farmacopees. De Wet op de Nederlandse Farmacopee (1871) legde vervolgens vast dat alle geneesmiddelen die in Nederland
verhandeld werden aan de eisen in de Nederlandse Farmacopee moesten voldoen en gaf haar zo kracht van wet.
Na de Tweede Wereldoorlog ontstond het idee om ook op Europese schaal de nationale farmacopees te harmoniseren. Gedurende de oorlog was namelijk een tekort aan geneesmiddelen ontstaan. Dat tekort had aangevuld kunnen worden met preparaten uit het buitenland, als men had geweten of die
buitenlandse producten uitwisselbaar waren met de bekende nationale producten. Bij gebrek aan gegevens over samenstelling van de buitenlandse producten en vanwege verwarring over de verschillende nationale productnamen werd echter maar weinig gebruikgemaakt van deze mogelijkheid.
Hierop volgde de gedachte een Europese Farmacopee samen te stellen met wetkracht in heel Europa. Op deze manier zou men erop kunnen vertrouwen dat de producten uit andere landen kwalitatief uitwisselbaar zijn met de nationaal bekende producten. Tegelijkertijd zou een Europese standaard bovendien de internationale handel in geneesmiddelen, en dus de beschikbaarheid van geneesmiddelen, bevorderen. De realisatie van de Europese Farmacopee werd mogelijk gemaakt in 1964 met de
opstelling en ondertekening van het Verdrag inzake de samenstelling van een Europese Farmacopee (zie Figuur 2.4).
2.1.2
Verdrag inzake de samenstelling van een Europese Farmacopee
In 1949 is de Raad van Europa (RvE) opgericht. Deze organisatie had en heeft de bevordering van samenhang van de deelnemende landen tot doel. Als onderdeel van ‘een sociale en goede
gezondheidszorg’ was en is het recht op kwalitatief goede en veilige geneesmiddelen een aandachtspunt van de Raad van Europa.
Iets later, in 1951, werd de voorloper van de Europese Unie, de Europese Gemeenschap voor Kolen en Staal, opgericht. Hieruit volgde in 1957 het Verdrag van Rome en de oprichting van de Europese Economische Gemeenschap (EEG). Deze Europese organisatie richtte zich op economische en politieke afspraken.
Politieke overwegingen hebben ertoe geleid dat de standaardisatie van kwaliteitseisen aan
geregistreerde geneesmiddelen is ondergebracht bij de Raad van Europa: alleen via deze organisatie konden de farmaceutisch belangrijke landen Verenigd Koninkrijk en Zwitserland bij het proces betrokken worden. In samenwerking met de organisatie van het Verdrag van Brussel (1951-1952), de West-Europese Unie (1953-1959) en de toenmalige EEG werd in 1964 het Verdrag inzake de
samenstelling van een Europese Farmacopee van kracht. In dit verdrag is vastgelegd dat de landen zich zullen inspannen tot het ontwikkelen en onderhouden van een Europese Farmacopee en dat men zorg zal dragen dat de Ph. Eur. ook nationaal geïmplementeerd wordt in de wetgeving. Dit verdrag is nog steeds van kracht en is voor het laatst herzien in 1992.(5)
In 1964 ondertekenden acht landen, waaronder Nederland, het Verdrag inzake de samenstelling van een Europese Farmacopee. Zie Bijlage 8 voor een weergave van de indrukken van dhr. Van Noordwijk, een Nederlandse vertegenwoordiger bij de Europese Farmacopee Commissie uit die tijd. Het betreft een partieel verdrag: de lidstaten van de Raad van Europa zijn niet verplicht het te ondertekenen maar kunnen zich op vrijwillige basis aansluiten. Het verdrag staat bovendien open voor ondertekening door partijen buiten de Raad van Europa.
Figuur 2.4 Voorpagina van het Verdrag omtrent de ontwikkeling van een Europese Farmacopee. (Verdragnummer 50, Straatsburg 22 juli 1964)
2.2
Huidig wettelijk kader
2.2.1
Europa
De farmacopee is begonnen als bundel bereidingsvoorschriften en kwaliteitseisen voor de bereiding van farmaceutische producten in de apotheek. Tegenwoordig bevat de Europese Farmacopee geen gedetailleerde bereidingsvoorschriften meer. De belangrijkste toepassing van de farmacopee betreft nu de kwaliteit van industrieel geproduceerde en nationaal of internationaal verhandelde producten.
Sinds 1975 is in EU Richtlijn 75/318/EEC vastgelegd dat de componenten van een geneesmiddel en het verpakkingsmateriaal moeten voldoen aan de monografieën in de Europese Farmacopee. Ook in de recentere Richtlijnen 2001/82/EC en 2004/82/EC voor veterinaire geneesmiddelen en Richtlijnen 2001/83/EC, 2003/63/EC en 2004/27/EC voor humane geneesmiddelen wordt verwezen naar de Europese Farmacopee. Richtlijn 2001/83/EC is van toepassing op homeopathische producten, kruidengeneesmiddelen, in de apotheek bereide, magistrale receptuur, bloed en bloedproducten, biologische producten, vaccins, immunologische serums en radiofarmaca. Het betreft verwijzingen op het gebied van benaming van grondstof, eindproduct, analysemethoden, specificatie, testapparatuur en normen, microbiologische eigenschappen en fabricageproces
Bovengenoemde Europese richtlijnen dienen op nationaal niveau geïmplementeerd te worden. In Nederland betekent dit dat in de Geneesmiddelenwet op diverse plaatsen naar de Europese Farmacopee verwezen wordt.
2.2.2
De Europese Farmacopee in mondiaal verband
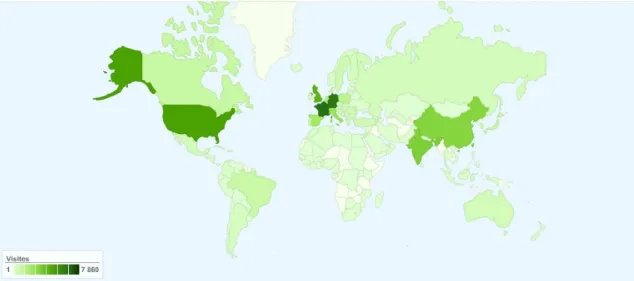
De zesde editie van de Europese Farmacopee is in 2008 verschenen in een totale oplage van 22.000 stuks en is zowel in boekvorm als digitaal beschikbaar. De abonnees zijn afkomstig uit 104 landen. De digitale versie van de farmacopee en achtergrondinformatie zijn toegankelijk via de website van het EDQM, www.edqm.eu. Het aantal maal dat deze website wordt geraadpleegd, stijgt jaarlijks. In 2007 werd de website per maand gemiddeld door 17.000 verschillende bezoekers geraadpleegd en het totaal aan bezoeken was gemiddeld 53.000 per maand.(6-7)
Figuur 2.5 Aantal bezoekers van de Ph. Eur. website in 2007, ingedeeld naar land (6)
Uit bovengenoemde gegevens is af te leiden dat de Europese Farmacopee wereldwijd gebruikt wordt bij het vaststellen van kwaliteitseisen voor geneesmiddelen. De primaire doelgroep van de Europese Farmacopee bestaat uit de spelers op de Europese geneesmiddelenmarkt. Maar ook de economisch geïnteresseerde landen van buiten de EU kunnen of moeten de Ph. Eur.-kwaliteit bij hun productie aanhouden. Internationale firma’s zijn aan de bepalingen van de Europese Farmacopee gehouden voor producten die zij in Europa voeren, ongeacht de locatie van het bedrijf.
De belangrijkste afnemers van de Europese Farmacopee buiten de EU en de RvE zijn, in afnemende volgorde: Verenigde Staten, India, China en Japan. Daarnaast wordt ook activiteit gesignaleerd in Canada, Australië, Brazilië en Argentinië.(6)
Naburige landen als Israël, Marokko en Tunesië, maar ook de landen die oorspronkelijk een kolonie van Europese landen waren zoals Australië, Canada en India hanteren vaak eisen die gebaseerd zijn op de Ph. Eur. In Tabel 2.1 is te zien dat deze landen dan ook waarnemer zijn bij de Europese Farmacopee Commissie. De gemenebestlanden Australië en Canada hebben de British Pharmacopoeia (BP) als nationale standaard aangenomen en dientengevolge is ook de Ph. Eur. in deze landen volledig van kracht.(8-9)
De officiële talen waarin de farmacopee verschijnt zijn Engels en Frans. Bij geschillen beroept men zich op de Franse tekst. Sinds juli 2006 stelt het European Directorate for the Quality of Medicines & HealthCare (EDQM) de Ph. Eur. ook in het Spaans beschikbaar. Deze versie betreft echter geen officiële vertaling. Op nationaal niveau wordt de farmacopee ook vertaald in bijvoorbeeld het Duits (samenwerking Oostenrijk, Zwitserland, Duitsland) en Portugees. Dergelijke vertalingen worden echter niet officieel erkend door de Raad van Europa en ook niet verspreid via het EDQM.
2.2.3
Nationale Farmacopees in Europa
Zoals vastgelegd in EU-Richtlijnen 2001/82/EC en 2001/83/EC heeft de Europese Farmacopee (Ph. Eur.) de hoogste status in Europa. Hierop volgen de nationale, Europese farmacopees en daarna overige farmacopees zoals de Amerikaanse (USP) of Japanse (JP).
Nederland heeft geen eigen nationale farmacopee meer. Een aantal andere Europese lidstaten echter nog wel: bijvoorbeeld het Verenigd Koninkrijk, Duitsland, Spanje en Frankrijk.
De Britse Farmacopee (BP) en de Franse Farmacopee (Ph. Fr.) onderscheiden zich van de Ph. Eur. door het opnemen van productmonografieën. In de BP zijn deze gebaseerd op de producten die in het Verenigd Koninkrijk op de markt zijn. Wat betreft de overige monografieën is deze farmacopee geen concurrent van de Ph. Eur.: deze worden letterlijk overgenomen.
Er zijn enkele monografieën in de Franse Farmacopee die niet in de Europese Farmacopee staan; voor bepaalde kleurpigmenten wordt bijvoorbeeld naar de Ph. Fr. verwezen en de Ph. Fr. bevat ook monografieën voor homeopathica. Duitsland onderhoudt in samenwerking met Oostenrijk en Zwitserland een Duitstalige farmacopee. Ook de Duitse farmacopee (Deutsches Arzneibuch, DAB) bevat nog enkele nationale monografieën. Daarnaast heeft Duitsland een aparte homeopathische farmacopee (Homeopathisches Arzneibuch, HAB). Spanje heeft ook nog een eigen farmacopee; de Ph. Es.
In Nederland heeft men in de praktijk weinig te maken met andere nationale farmacopees, omdat de relevante monografieën uit de diverse lidstaten via een procedure van Adaptation of National
Monographs (ANM), indien relevant bevonden, reeds zijn overgenomen in de Europese Farmacopee.
2.2.4
Overige, internationale farmacopees
In internationaal verband zijn de drie grootste farmacopees de Europese Farmacopee (Ph. Eur.), de United States Pharmacopeia (USP) en de Japanese Pharmacopoeia (JP). Deze omvatten samen het grootste deel van de mondiale geneesmiddelenmarkt. De organisatie achter deze drie farmacopees loopt echter sterk uiteen en ook de reikwijdte van de opgenomen kwaliteitseisen verschilt.
De USP is een private, Amerikaanse instelling die een Engelstalige en Spaanstalige farmacopee uitgeeft. De USP heeft geen winstoogmerk. Gelden worden verkregen uit verkoop van producten en diensten in het kader van farmaceutische zorg. De USP wordt net als de Ph. Eur. ondersteund door vrijwilligers uit industrie, overheid, en consumentenorganisaties. Zie de website www.usp.org voor meer informatie.
De USP heeft geen wetkracht en is niet direct verbonden met de Amerikaanse registratieautoriteit FDA (Food and Drug Administration). Het is dan ook niet vanzelfsprekend dat de eisen die opgenomen zijn in de USP goedgekeurd worden door de FDA. Dit in tegenstelling tot de eisen in de Ph. Eur. die wetkracht hebben en dus door registratieautoriteiten gehanteerd dienen te worden. De arbitragefunctie van de USP bij registratiediscussies is als gevolg hiervan minder uitgesproken dan die van de Ph. Eur. USP en Ph. Eur. concurreren wat betreft de toepassing binnen de Spaanstalige Amerikaanse landen en staten. Sinds kort wordt echter ook de Europese Farmacopee (door Spanje) vertaald in het Spaans. Ook is een Portugese versie verkrijgbaar. Deze versies van de farmacopee zijn weliswaar niet officieel erkend, maar zij maken de Ph. Eur. wel toegankelijk voor landen in Midden- en Zuid-Amerika. De JP wordt door de Japanse overheid opgesteld. Op de website www.sjp.jp is meer informatie te vinden over de Society of Japanese Pharmacopoeia (SJP). Deze non-profitorganisatie valt onder het Japanse ministerie van Gezondheid, Arbeid en Welzijn.
In verhouding lijkt de organisatie van de Ph. Eur. flexibeler georganiseerd dan die van concurrenten USP en JP. Procedureel wijkt de Ph. Eur. bijvoorbeeld af van USP en JP door landen van buiten Europa de mogelijkheid te bieden om een waarnemerstatus aan te vragen. Het EDQM presenteert zichzelf ook als een organisatie die ernaar streeft voorop te lopen wat betreft actualiteit, flexibiliteit en innovatie. Over concurrentie met de Japanse Farmacopee is geen informatie gevonden.
De Chinese Farmacopee is in opkomst vanwege de uitbreiding van de farmaceutische markt in China. Door de start van veel grondstoffabrikanten in China worden steeds meer grondstoffen uit China betrokken. Het is voor een grote en groeiende economie als de Chinese, van groot belang om de ontwikkelingen in Europa nauwgezet te volgen. China is dan ook waarnemer bij de Europese
Farmacopee Commissie. Het EDQM stimuleert deze contacten zoals te lezen valt in het jaarverslag van het EDQM over 2007.(7)
Andere nationale farmacopees waarmee internationaal opererende firma’s te maken hebben zijn bijvoorbeeld de Indiase, de Russische en de Braziliaanse. De Indiase Farmacopee baseert zich grotendeels op de Britse Farmacopee; indirect bestaat zo een verband met de Europese Farmacopee. Een voorbeeld van een land dat in het verleden voornemens was de eigen farmacopee te onderhouden is de Russische Federatie. Sinds juni 2006 is dit land echter ook waarnemer bij de Europese
Farmacopee Commissie. Ook Kazakstan heeft de status van waarnemer verkregen. Deze twee landen zullen mogelijk op termijn ook de Europese Farmacopee gaan hanteren.
De Wereldgezondheidsorganisatie (WHO) geeft ook een farmacopee uit: de zogenaamde Internationale Farmacopee. Deze International Pharmacopoeia (IntPh) is met name gericht op de medicatie die in ontwikkelingslanden gebruikt wordt en bevat IntPh-kwaliteitsspecificaties en analysemethoden voor farmaceutische producten, hulpstoffen en toedieningsvormen. In tegenstelling tot de Europese
Farmacopee, heeft de Internationale Farmacopee geen wettelijke status. Meer informatie is te vinden op de website van de WHO: www.who.int.
2.2.5
Internationale harmonisatie
Voor een internationaal opererende firma is het een complexe en tijdrovende aangelegenheid om aan alle bepalingen van de drie (of meer) belangrijke farmacopees te voldoen. Soms moet één type onderzoek op drie verschillende manieren uitgevoerd worden om wereldwijd geaccepteerd te worden. Dit geldt bijvoorbeeld op dit moment voor het onderzoek op steriliteit. Harmonisatie van de drie meest gebruikte farmacopees (Ph. Eur., USP en JP) is daarom wenselijk vanuit het perspectief van de industrie.
Aan deze harmonisatie wordt gewerkt in de Pharmacopoeial Discussion Group (PDG) waarin vertegenwoordigers van Ph. Eur., USP, JP en WHO zitting hebben. De organisatie van de Europese Farmacopee is gebaseerd op samenwerking tussen de betrokken Europese lidstaten. Deze ervaring met harmonisatieprocessen kan gunstig ingezet worden in de PDG.
De eerste successen zijn inmiddels geboekt. De methode voor het bepalen van asrestbepaling van deze drie farmacopees is bijvoorbeeld geharmoniseerd. Ook de testen voor bepaling van microbiële
contaminatie van niet-steriele producten (hoofdstukken 5.1.4, 2.6.12 en 2.6.13 van de Ph.Eur.) zijn geharmoniseerd per 1 januari 2007. Dit soort harmonisatie mag op het oog triviaal lijken, voor een farmaceutische firma voorkomt het echter zeer veel onnodige en tijdrovende arbeid.
Harmonisatie op deelgebieden vindt ook plaats en wordt harmonisation by attribute genoemd. In geharmoniseerde monografieën wordt aangegeven wanneer er een gedeelte buiten de harmonisatie valt. In de Verenigde Staten wil men bijvoorbeeld voor grondstoffen een test op residuele oplosmiddelen die kunnen voorkomen na het synthetiseren van de grondstof, door contact met verontreinigingen tijdens transport. De Ph. Eur. neemt dit type eisen niet over, omdat transport niet beschouwd wordt als onderdeel van het productieproces.
In de toekomst kan harmonisatie met andere farmacopees ook een rol gaan spelen bijvoorbeeld met die van Brazilië, China of India. Uit het jaarverslag 2007 van het EDQM blijkt ook dat er diverse contacten zijn met andere internationale autoriteiten (7).
2.2.6
De inhoud van Europese Farmacopee in 2008
De Europese Farmacopee bevat monografieën voor grondstoffen, producten en verpakkingsmaterialen. Deze monografieën hebben wetkracht voor de producten waarop zij van toepassing zijn. De huidige, zesde editie van de farmacopee bevat ruim tweeduizend monografieën.
Een monografie beschrijft de testen en ook de eraan gerelateerde eisen waarmee de kwaliteit van de grondstof of het product getoetst kan worden. Daarnaast zijn verwijzingen naar apparatuur en
referentiestandaarden opgenomen. Op deze manier is een openbaar totaalpakket beschikbaar waarvan iedereen gebruik kan maken die betrokken is bij bereiding, registratie en controle van geneesmiddelen. Een dergelijk pakket kan van belang zijn in geval van geschillen: uitslagen kunnen volgens de Ph. Eur. nagewerkt worden door een onafhankelijke derde (arbitrage).
Het eerste hoofdstuk ‘General Notices’ in de farmacopee beschrijft de structuur van een monografie. De ‘Technical Guide for elaboration of monographs’ van het EDQM beschrijft hoe de verschillende tests in een monografie tot stand kunnen komen.(10)
De grondstofmonografieën betreffen zowel werkzame bestanddelen als hulpstoffen van chemische, biologische of semi-synthetische oorsprong. De farmacopee beschrijft ook algemene eisen voor toedieningsvormen zoals tabletten of inhalatiepreparaten maar ook voor bijvoorbeeld
radiofarmaceutica, vaccins en kruidenthee. Voor de biologische producten vaccins en sera zijn daarnaast specifieke productmonografieën opgenomen.
Naast monografieën zijn algemene teksten opgenomen die de monografieën overstijgen en, indien van toepassing, in acht dienen te worden genomen. Deze teksten gaan bijvoorbeeld over steriliteit, residuele oplosmiddelen en het gebruik van referentiestandaarden.
In de Ph. Eur. zijn minimale kwaliteitseisen voor diverse productaspecten opgenomen. De vastgestelde eisen zijn gebaseerd op de beoogde toepassing en houden rekening met zowel de veiligheid voor de gebruiker als met technische haalbaarheid. Het veilige gebruik van een product wordt gewaarborgd door het vastleggen van limieten voor onder meer potentieel toxische onzuiverheden. De eisen worden gerelateerd aan de kwaliteit van producten die reeds zijn geregistreerd op de Europese markt en aan actuele wetenschappelijke kennis. De farmaceutisch-technische eisen die aan producten en
verpakkingsmaterialen gesteld worden, zijn bedoeld om een minimale kwaliteit van de producten te garanderen en te zorgen dat de gebruiker een product ontvangt dat geschikt en veilig is voor de beoogde toepassing.
Figuur 2.6 Voor de entree van de Raad van Europa: de diverse nationale vlaggen met op de voorgrond een gedicht uit Nederland (‘Denkend aan Holland’ van H. Marsman) ter gelegenheid van het Nederlandse voorzitterschap van de Raad van Europa van 6 november 2003 tot 12 mei 2004 (foto: D. de Kaste).
2.3
Huidig organisatorisch kader
2.3.1
Raad van Europa
De Raad van Europa is in 1949 opgericht door 8 landen met als doel eenheid te bevorderen tussen de leden. Op dit moment bestaat de Raad van Europa uit 47 landen, waaronder alle EU-lidstaten. Alle landen bij elkaar omvatten ongeveer 800 miljoen Europeanen. Het besluitvormende orgaan van de Raad van Europa wordt gevormd door de ministers van Buitenlandse Zaken en hun
vertegenwoordigers. Deze Commissie van Ministers besluit onder andere over het budget van de raad. Het totale budget voor 2006 bedroeg 262 miljoen euro (11).
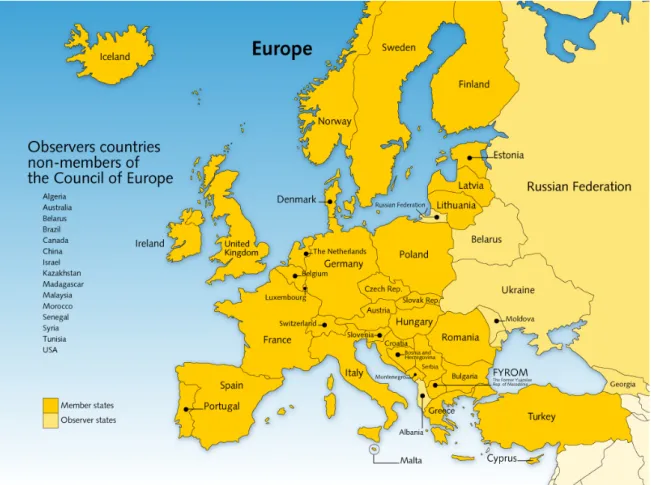
Figuur 2.7 Leden en waarnemende landen van de Raad van Europa in december 2008 (13)
De Parlementaire Vergadering vertegenwoordigt de regeringen van de lidstaten. Het aantal
vertegenwoordigers en de financiële bijdrage van elk land is vastgesteld op basis van inwoneraantal en rijkdom. Het parlement van de Raad van Europa bestond in maart 2008 uit 318 nationale
vertegenwoordigers en hun vervangers. Het aantal vertegenwoordigers per lidstaat varieert van 2 tot 18; Nederland heeft 7 vertegenwoordigers in het parlement. Voor meer informatie, zie www.coe.int. De officiële taal van de Raad van Europa is Frans. De Europese Farmacopee is officieel beschikbaar in zowel de Franse als de Engelse taal. De Raad van Europa verschilt hierin van de Europese Unie, waar officiële documenten in principe in alle Europese talen beschikbaar worden gemaakt. Dit kost de EU veel geld en tijd (12). Hiervan is bij de Raad van Europa veel minder sprake. In de praktijk wordt behalve in het Frans veel in het Engels gewerkt.
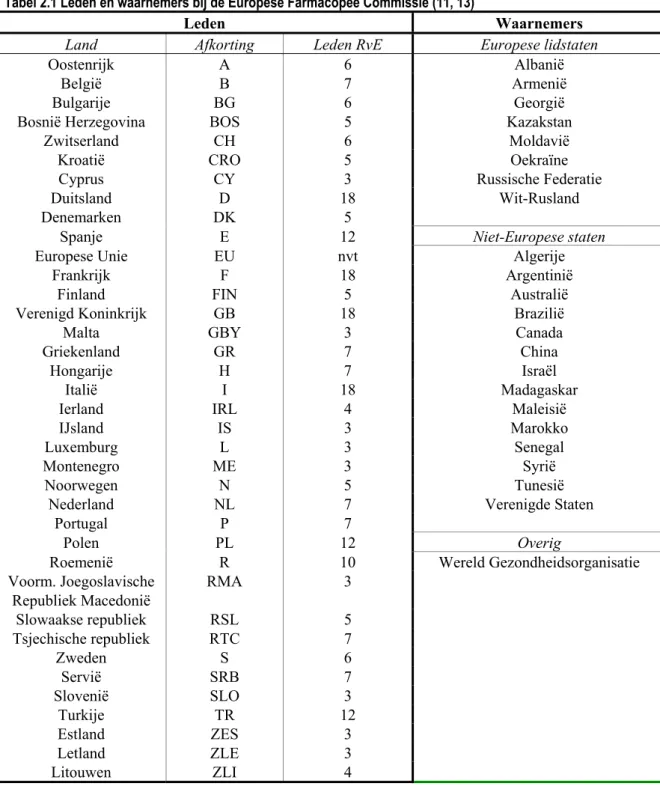
Tabel 2.1 Leden en waarnemers bij de Europese Farmacopee Commissie (11, 13)
Leden Waarnemers
Land Afkorting Leden RvE Europese lidstaten
Oostenrijk A 6 Albanië
België B 7 Armenië
Bulgarije BG 6 Georgië
Bosnië Herzegovina BOS 5 Kazakstan
Zwitserland CH 6 Moldavië
Kroatië CRO 5 Oekraïne
Cyprus CY 3 Russische Federatie
Duitsland D 18 Wit-Rusland
Denemarken DK 5
Spanje E 12 Niet-Europese staten
Europese Unie EU nvt Algerije
Frankrijk F 18 Argentinië
Finland FIN 5 Australië
Verenigd Koninkrijk GB 18 Brazilië
Malta GBY 3 Canada
Griekenland GR 7 China
Hongarije H 7 Israël
Italië I 18 Madagaskar
Ierland IRL 4 Maleisië
IJsland IS 3 Marokko
Luxemburg L 3 Senegal
Montenegro ME 3 Syrië
Noorwegen N 5 Tunesië
Nederland NL 7 Verenigde Staten
Portugal P 7
Polen PL 12 Overig
Roemenië R 10 Wereld Gezondheidsorganisatie
Voorm. Joegoslavische Republiek Macedonië RMA 3 Slowaakse republiek RSL 5 Tsjechische republiek RTC 7 Zweden S 6 Servië SRB 7 Slovenië SLO 3 Turkije TR 12 Estland ZES 3 Letland ZLE 3 Litouwen ZLI 4
Het (partiële) verdrag omtrent de Europese Farmacopee was in juli 2008 ondertekend door 37 van de 47 leden van de Raad van Europa, zie Tabel 2.1 (13). Montenegro is geen lid van de raad maar is wel een verdragspartij. Daarnaast heeft de EU als internationale organisatie het verdrag geratificeerd. In de tabel is tevens aangegeven hoeveel vertegenwoordigers het betreffende land heeft in de parlementaire
vergadering van de Raad van Europa. Hieruit kan een indruk worden verkregen van de inspraak van het land in de totale Parlementaire Vergadering van de Raad van Europa.
Uit Tabel 2.1 blijkt verder dat 23 partijen de status van waarnemer hebben, waaronder de WHO. De ‘ontbrekende’ Europese landen zijn Andorra, Azerbeidzjan, Liechtenstein, Malta, Moldavië, Monaco, San Marino, Staat Vaticaanstad. Voor de meeste van deze landen geldt echter dat zij zich richten op grotere (buur)landen wat betreft de kwaliteitsbeoordeling van geneesmiddelen.
De organisatie rond de Europese Farmacopee vindt plaats in Straatsburg, de standplaats van de Raad van Europa. Hier beheert het European Directorate for the Quality of Medicines & Health Care (EDQM) het technische secretariaat van de Europese Farmacopee en vinden de vergaderingen van de Europese Farmacopee Commissie plaats. In de volgende paragrafen worden deze twee organisaties toegelicht.
2.3.2
Europese Farmacopee Commissie
De Europese Farmacopee Commissie is formeel het belangrijkste orgaan bij het tot stand komen van de Ph. Eur. In het Verdrag op de Europese Farmacopee is vastgelegd dat de Ph. Eur. onder de
verantwoordelijkheid valt van de Public Health Committee (Volksgezondheidscommissie) van de Raad van Europa. De Europese Farmacopee Commissie is samengesteld om het voorbereidende werk te doen; zij adviseert en rapporteert aan deze Volksgezondheidscommissie.
De taken van de Europese Farmacopee Commissie zijn vastgelegd in artikel 4 van de huidige Conventie. Hierin staat dat de commissie algemene voorwaarden voor het onderhouden van de Europese Farmacopee vaststelt. De commissie bepaalt het werkprogramma van de expert- en
werkgroepen. De commissie draagt zorg voor het voorbereiden en aannemen van nieuwe monografieën voor opname in de Europese Farmacopee en zij neemt besluiten omtrent analytische methoden die gehanteerd worden. Tot slot doet zij aanbevelingen voor het vaststellen van tijdslimieten waarbinnen technische aanpassingen in de Europese Farmacopee geïmplementeerd dienen te worden door de betrokken lidstaten.
De Europese Farmacopee Commissie wordt gevormd door alle verdragspartijen en een
vertegenwoordiger van de EU Pharmaceuticals Unit DG Enterprise. Omdat de EU de Conventie over de Europese Farmacopee heeft ondertekend namens alle EU-landen, voert de EU-vertegenwoordiger voor bestuurlijke beslissingen het woord namens alle EU-lidstaten. Wat betreft technische zaken worden de beslissingen door de afgevaardigden van de individuele landen genomen. Elk land mag met maximaal drie afgevaardigden deelnemen aan de vergadering, maar heeft altijd maar één stem.
Daarnaast telt de Commissie twintig leden met de status van waarnemer waaronder vertegenwoordigers van de European Medicines Agency (EMEA) en de WHO.
De vergadering wordt voorgezeten door een presidium bestaande uit een voorzitter en twee vice-voorzitters. De gang van zaken rond en tijdens de vergadering ligt vast in de Rules of procedure (14). De Europese Farmacopee Commissie staat in contact met de nationale overheden van de betrokken lidstaten. In Nederland bepaalt het ministerie van VWS de nationale bijdrage aan de Europese
Farmacopee. Zo wordt in overleg tussen de commissie en het ministerie van VWS de begroting van het EDQM besproken en de Nederlandse inbreng hierin vastgesteld.
2.3.2.1 Onderhoud en aanvulling Europese Farmacopee
De farmacopee is niet statisch, maar wordt aangepast op basis van nieuwe technische ontwikkelingen en inzichten. Zie bijvoorbeeld Bijlage 2 voor de ontwikkeling van de monografie van Morfine in de loop der tijd.
Het doorlopende werkprogramma voor het onderhouden en aanvullen van de farmacopee wordt tijdens de vergadering steeds opnieuw bijgesteld door de Europese Farmacopee Commissie. (EPC). Het proces start met het aanwijzen van de te ontwikkelen monografieën. De benodigde inspanningen worden geleverd vertegenwoordigers van de verdragspartijen. De Europese Farmacopee Commissie wordt
hiertoe ondersteund door experts en specialisten die in groepen werken aan voorstellen voor nieuwe en aangepaste farmacopeeteksten.
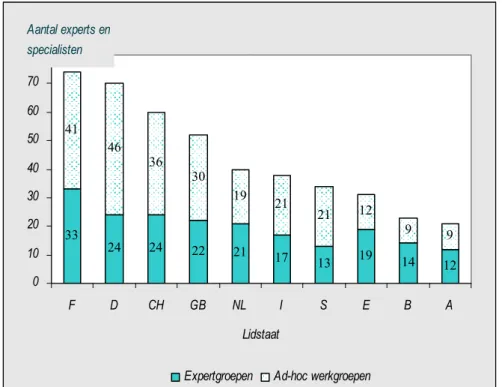
In december 2007 waren er 18 permanente expertgroepen (15). Daarnaast waren er 35
ad-hocwerkgroepen die kortdurend over een specifiek onderwerp adviseren. De experts en specialisten voor de werkgroepen worden voorgedragen door de nationale delegaties en voor 3 jaar benoemd door de Europese Farmacopee Commissie. Deelnemers zijn afkomstig van industrie, universiteiten, controlelaboratoria, inspectie en registratieautoriteiten.
De vergaderfaciliteiten van de expertgroepen worden beschikbaar gesteld door het EDQM (zie paragraaf 2.3.3). Over het algemeen worden reis- en verblijfskosten van experts vergoed uit het
nationale budget. De laboratoriumondersteuning en inzet van ondersteunend personeel is voor rekening van de expert. Experts en specialisten van bedrijven gaan meestal op eigen kosten.
Zodra een conceptversie van een nieuwe of herziene tekst beschikbaar is, wordt deze gepubliceerd in het tijdschrift Pharmeuropa. Dit tijdschrift wordt door het EDQM toegestuurd aan abonnees en is ook digitaal toegankelijk. Belanghebbenden worden daarmee in de gelegenheid gesteld commentaar te leveren op de voorgestelde teksten. Commentaar dient binnen drie maanden na publicatie te worden ingestuurd.
De nationale delegatie draagt er zorg voor dat het vanuit Nederland geleverde commentaar het secretariaat van de Ph. Eur. bereikt. Zij moedigt belanghebbenden aan ideeën voor verbetering aan te dragen en vertegenwoordigt het Nederlandse belang bij het secretariaat. Het contact met het EDQM kan ook verlopen via koepelorganisaties die commentaar verzamelen overdragen aan de nationale delegatie of direct indienen bij het EDQM. Belanghebbenden uit landen die het verdrag op de Europese Farmacopee niet hebben ondertekend, kunnen commentaar rechtstreeks naar het EDQM sturen.
2.3.2.2 Ontwikkelingsproces
Na het vaststellen van de werklijst door de Europese Farmacopee Commissie, wordt één van de experts uit de betreffende expertgroep of ad-hocwerkgroep dan aangesteld als ‘rapporteur’ voor de betreffende monografie. De tijd die de rapporteur nodig heeft voor het ontwikkelen van een conceptmonografie is afhankelijk van de beschikbare informatie. Wanneer er een monografie beschikbaar is in bijvoorbeeld de USP, de JP of andere farmacopees kan deze als basis gebruikt worden. Ook kan informatie van de grondstoffabrikant over relevante aspecten, analysemethoden en haalbare limieten als basis gebruikt worden. Naast het opstellen van een conceptmonografie worden de methoden, met name de test op verwante verbindingen, ook nog nagewerkt, geoptimaliseerd en gevalideerd. Tevens worden de limieten gezet aan de hand van resultaten van diverse batches op de EU-markt. Naar schatting kan de tijd voor het opstellen van een conceptmonografie variëren van 60 tot 200 uur.
De conceptmonografie wordt vervolgens voorgelegd aan de rest van de expertgroep voor commentaar. Na verwerking van geleverd commentaar stelt de groep een concept op dat gepubliceerd wordt in Pharmeuropa. De vergadertijd die hieraan besteed in een expert groep wordt is naar schatting per monografie 1 à 2 uur.
Om de robuustheid van de voorgestelde analytische methoden te toetsen wordt de conceptmonografie door diverse belanghebbenden nagewerkt op de belangrijkste punten. In Nederland gebeurt dit onder andere door de leden van de Werkgroep Monografie Evaluatie. Naar schatting kost het nawerken van de belangrijkste tests van een monografie gemiddeld 60 uur. Voor uitgebreidere monografieën of complexere stoffen zal dit uiteraard hoger liggen.
Het commentaar dat volgt op de publicatie in Pharmeuropa wordt verzameld, besproken in de expertgroep en indien van toepassing verwerkt in een nieuwe conceptmonografie. Dit tweede concept wordt ter goedkeuring voorgelegd aan de Europese Farmacopee Commissie. Indien de Commissie niet akkoord gaat, wordt de monografie teruggestuurd naar de expertgroep. Indien de Commissie de monografie als ‘definitieve versie’ aanneemt, volgt implementatie door publicatie van de nieuwe tekst in de Europese Farmacopee.
2.3.2.3 Aanpassingen in het proces
Het proces rond het bijhouden van de Europese Farmacopee wordt steeds herzien en aangepast om de efficiëntie en snelheid waarmee geactualiseerde en nieuwe monografieën verschijnen te verhogen (16-17). De efficiëntie van de ontwikkeling van nieuwe teksten is verhoogd door inzet van meer mensen en versoepeling van het werkproces. Er zijn in korte tijd veel nieuwe monografieën aangenomen; in 2005 zijn bijvoorbeeld 168 nieuwe en herziene monografieën vastgesteld. Dit heeft onder meer te maken met het versneld overnemen van monografieën uit nationale farmacopees. Deze monografieën waren immers al nationaal van kracht en dienden alleen nog tekstueel en qua lay-out te worden aangepast. De taken van de hiertoe aangestelde commissie voor Adaptation of National Monographs zijn inmiddels afgerond. Daarnaast is het mogelijk gemaakt om monografieën gedeeltelijk aan te nemen en na implementatie verder te werken aan optimaliseren ervan. Dit proces heeft ertoe geleid dat het belang van de nationale farmacopees is verminderd en een breder Europees kader is geschapen.
De inhoud van de monografieën is aangepast om beter aan te sluiten op de praktijk van de gebruikers. Dit geldt onder meer wat betreft de opgenomen informatie over onzuiverheden. Voor de duidelijkheid worden nu de structuurformules van de bekende onzuiverheden vermeld in de monografie
(zie Bijlage 3 voor de huidige monografie voor Morfine bijvoorbeeld).
2.3.2.4 Toekomst
Het werkprogramma van de Europese Farmacopee wordt elke vergadering opnieuw vastgesteld. Daarnaast houdt de commissie rekening met actuele zaken en ontwikkelingen in de farmacie. Op de EDQM-website zijn zogenoemde ‘hot topics’ terug te vinden waarmee rekening is en wordt gehouden bij de ontwikkeling van editie 6 (2007) van de Europese Farmacopee: special revision program (herzien van monografieën op het gebied van limieten voor onzuiverheden), eigenschappen van hulpstoffen die van belang kunnen zijn voor verwerking in het eindproduct (functionality related characteristics, bijvoorbeeld deeltjesgrootte) en kruidenpreparaten.
Het EDQM organiseert symposia over actuele zaken (bijvoorbeeld herziening van de monografie van heparine in 2008) en ook over onderwerpen waar men in de toekomst de farmacopee mogelijk op wil laten aansluiten. Zo was er op 20 juni 2007 bijvoorbeeld een symposium over kwaliteitsstandaarden voor geneesmiddelen die niet geregistreerd zijn, zoals ad-hocbereidingen in de apotheek, kleinschalige bereidingen in een ziekenhuisapotheek of radiofarmaceutica. Meer informatie over dit symposium en ook over andere activiteiten van het EDQM is te vinden via www.edqm.eu. Als vervolg op
bovengenoemd symposium wordt nu gewerkt aan de samenstelling van een werkgroep over dit type producten.
Een andere ontwikkeling in de farmacie is de trend om processen te controleren in plaats van het eindproduct. De Ph. Eur. is nog niet ingericht op de gevolgen van documenten als ICH Q8
Pharmaceutical Development, Q9 Quality Risk Management en Q10 Pharmaceutical Quality System. In de toekomst zal echter mogelijk een andere manier van controle nodig zijn dan de huidige manier van eindcontroles die beschreven worden in de huidige Ph. Eur.-monografieën. De Europese Farmacopee Commissie acht het van belang dat de Ph. Eur. op dit gebied betrokken blijft bij de ontwikkelingen. Er wordt daarom gewerkt aan de oprichting van een werkgroep Process Analytical Technologies (PAT), die zich bezig kan houden met technieken die geschikt zijn om processen volgens de nieuwe trend te controleren.
2.3.3
EDQM and HealthCare
Het European Directorate for the Quality of Medicines & Health Care (EDQM) in Straatsburg beheert het technische secretariaat van de Europese Farmacopee. Dit secretariaat bestaat al sinds het Verdrag op de Europese Farmacopee en werd aanvankelijk Europese Farmacopee Secretariaat genoemd. In
1995 werd het EDQM (toen: European Department for the Quality of Medicines) opgericht. Haar takenpakket omvatte naast het Ph. Eur.-secretariaat ook gerelateerde activiteiten zoals het
certificeringsschema en de coördinatie van het Europese netwerk van controle laboratoria (OMCL-netwerk). Via het EDQM wordt ook standaardisatie van biologische standaarden mogelijk gemaakt via het Biological Standardisation Program.
Het takenpakket van het EDQM is per 1 januari 2007 uitgebreid met bloedproducten en
celtherapeutica, vandaar de toevoeging ‘and Health Care’ aan haar naam. Meer informatie hierover is te vinden op de website van het EDQM (www.edqm.eu). Hieronder worden enkele van de taken van het EDQM nader toegelicht.
Figuur 2.8 Het EDQM-gebouw in Straatsburg: overzicht en entree (foto’s: G. Thole)
De kosten die het EDQM maakt voor het beschikbaar stellen van de vergaderfaciliteiten van de expertgroepen zijn niet exact bekend. Wel is duidelijk dat het EDQM in 2007 20% van de totale begroting financierde met nationale bijdragen. Dit omvat niet alleen de kosten voor het secretariaat van de Europese Farmacopee Commissie maar ook alle overige taken. De voorlopige begroting van 2007 vermeldde dat de Nederlandse overheid ongeveer 4% van deze 20% bijdraagt; dit zou overeenkomen met ongeveer 132.000 euro (18). Teruggerekend zou dit betekenen dat de begroting van het EDQM in de orde van 16 à 17 miljoen euro ligt. De overige financiering bestaat onder meer uit budget van de Raad van Europa, bijdragen van andere belanghebbende partijen aan de activiteiten (zoals de EU) en inkomsten van het EDQM zelf. Het EDQM genereert immers inkomsten door de wereldwijde verkoop van de Europese Farmacopee en van referentiestandaarden. Hoewel de cijfers niet beschikbaar zijn, mag aangenomen worden dat deze inkomsten positief bijdragen aan het functioneren van het EDQM en aan de financiële positie van het EDQM binnen de Raad van Europa.
2.3.3.1 Secretariaat Europese Farmacopee Commissie
Het EDQM vormt de spil in de contacten tussen nationale delegaties, industriële partijen en de Europese Farmacopee. Wat Nederland betreft draagt de nationale delegatie over het algemeen zorg voor de contacten met het secretariaat van de Ph. Eur., bijvoorbeeld wat betreft het vanuit Nederland geleverde commentaar op conceptmonografieën in Pharmeuropa. Industriële koepelorganisaties kunnen echter ook direct contact opnemen met het EDQM. Het EDQM is ten slotte ook direct te benaderen door belanghebbenden uit landen die het verdrag op de Europese Farmacopee niet hebben ondertekend.
2.3.3.2 Certificatieschema en EDQM-inspecties
In Richtlijn 2001/83/EC wordt verwezen naar de mogelijkheid van het certificeren van grondstoffen. Fabrikanten van grondstoffen die in de Ph. Eur. zijn opgenomen, kunnen een dossier indienen bij het EDQM waarin het productieproces en de specificatie van de door hen geproduceerde stof worden beschreven. Aan de hand van dit dossier wordt in Straatsburg beoordeeld of de Ph. Eur.-monografie geschikt is om de kwaliteit van de betreffende stof te controleren. Bij positief resultaat wordt een Certificate of Suitability of the European Pharmacopoeia (CEP) afgegeven waar dit op staat vastgelegd. Waar nodig, worden op het certificaat ook vereiste aanvullende tests en eisen vermeld; de te gebruiken methoden worden dan bijgevoegd (CEP+). Indien relevant, kan hieruit ook een voorstel tot revisie van de monografie voortkomen. Op aanvraag van de fabrikant kunnen ook de houdbaarheid en het
verpakkingsmateriaal op het certificaat worden beoordeeld en vastgelegd. Sinds de invoering van de certificatieprocedure zijn meer dan 2380 certificaten toegekend (7).
Deze certificaten worden erkend door de registratieautoriteiten. Wanneer de registratieaanvrager in het registratiedossier een kopie opneemt van het certificaat, hoeft het dossiergedeelte dat gedekt wordt door het certificaat niet opnieuw beoordeeld te worden. De beoordelingstijd van de autoriteiten wordt hierdoor verkort en het registratieproces versneld.
Ten gevolge van het certificatieschema heeft het EDQM toegang gekregen tot en kennis van de inhoud van registratiedossiers van werkzame bestanddelen. Ter controle van de papieren data voert zij
bedrijfsinspecties uit. Zowel grondstoffabrikanten als tussenhandelaren moeten voor het verkrijgen van een certificaat aangeven bereid te zijn een Good Manufacturing Practice (GMP)-inspectie te ondergaan. In 2007 hebben 34 inspecties plaatsgevonden, voornamelijk in India en China (7). Deze inspecties vinden plaats aanvullend op die van de nationale Europese autoriteiten, dat wil zeggen op plaatsen waar deze niet komen. Verder is samenwerking gezocht met andere instanties en landen buiten Europa (Australië). Deze Europese bijdrage aan inspecties van grondstofleveranciers is des te actueler omdat het toezichtregime voor grondstofproductie recent is aangescherpt en meer inspecties vereist. De inspecties vanuit het EDQM kunnen dus ook veel nieuwe nationale inspanningen ondervangen.
2.3.3.3 Normalisatie nomenclatuur
In de Europese Farmacopee wordt de naamgeving van werkzame bestanddelen, hulpstoffen en reagentia eenduidig vastgelegd. Waar mogelijk wordt de nomenclatuur in overeenstemming gebracht met de International Nonproprietary Names (INN) zoals deze door de WHO zijn gedefinieerd. Deze nomenclatuur voorkomt misverstanden over de identiteit van de stoffen waar registratiemedewerkers bij overheid en industrie mee te maken hebben.
De monografieën voor plantenmateriaal zijn weliswaar nog niet groot in aantal, maar ook voor die producten heeft de Europese Farmacopee een rol bij de definiëring van herkomst, samenstelling en daaraan gekoppelde nomenclatuur. Vanwege de voorkeurstalen Frans en Engels is hierbij het voor de hand liggende gebruik van Latijnse benamingen echter naar de achtergrond verdwenen. Dit leidt soms tot verwarring. Botanische herkenning wordt met tekeningen mogelijk gemaakt.
Naast de Europese Farmacopee geeft het EDQM ook een lijst met standaardtermen uit voor farmaceutische toedieningsvormen, toedieningswegen en verpakkingsmaterialen. Deze lijst is ontwikkeld op verzoek van de EMEA, vanwege de verwarring veroorzakende uiteenlopende naamgeving van producten. In Europa is de industrie nu verplicht zich aan deze vastgelegde nomenclatuur te houden. Dit bevordert de eenduidigheid van de gebruikte termen in Summary of Product Characteristics (SPC), bijsluiters en op de verpakking.
2.3.4
Europese Unie
Vanuit beleidsoptiek hebben geneesmiddelen een tweeledig karakter: enerzijds zijn het
handelsproducten met een economisch belang, anderzijds zijn het producten waar de volksgezondheid vanaf hangt. In de regelgeving rond geneesmiddelen is dit tweeledige karakter ook te herkennen. De
sociale aspecten zijn voor de Raad van Europa reden zich bezig te houden met geneesmiddelen. De economische belangen met betrekking tot handelsvergunningen worden door de Europese Unie geregeld.
Uit de Richtlijnen 2001/82/EC en 2001/83/EC volgt dat de Europese Farmacopee van toepassing is op alle aanvragen voor een handelsvergunning van een geneesmiddel in de EU. En ook voor
niet-geregistreerde producten gelden de eisen van de Europese Farmacopee. Dit betekent dat de Europese Farmacopee de kwaliteit van de geneesmiddelen die beschikbaar zijn voor 450 miljoen Europeanen bewaakt (19).
De wet- en regelgeving omtrent toelating en het toezicht op geneesmiddelen binnen de EU is ondergebracht bij de afdeling DG Enterprise. De EU houdt zich bezig met het opstellen van geharmoniseerde registratie-eisen om de handelsbarrières voor geneesmiddelen te beperken. De European Medicines Agency (EMEA) te Londen is het Europese instituut dat handelsvergunningen verleent aan geneesmiddelen die de centrale registratieprocedure hebben doorlopen. Zie ook http://europa.eu/index_nl.htm en www.emea.eu.int (19-20).
Het Committee for Medicinal Products for Human Use (CHMP) van de EMEA is het wetenschappelijk ondersteunende orgaan voor geneesmiddelen voor humaan gebruik. Het Committee for Medicinal Products for Veterinary Use (CVMP) doet hetzelfde voor producten voor veterinair gebruik. Wat betreft de kwaliteit van geneesmiddelen beschikken de CHMP en CVMP over verschillende werkgroepen, waaronder de Joint CHMP/CVMP Quality Working Party (QWP) en, alleen voor de CHMP, de Biologics Working Party (BWP), maar ook de Blood Products Working Party (BPWP) en de Vaccine Working Party (VWP). Deze werkgroepen zijn samengesteld uit vertegenwoordigers van de nationale lidstaten. Zij stellen onder meer richtsnoeren op voor de ontwikkeling en registratie van geneesmiddelen.
De EU neemt deel aan vergaderingen van de Europese Farmacopee Commissie. Op haar beurt is ook de RvE vertegenwoordigd bij vergaderingen van de EU. Men streeft ernaar de overlap van de activiteiten die de EU uitvoert en de initiatieven van de RvE/EDQM zo beperkt mogelijk te houden.
2.3.5
Ontwikkeling, registratie, keuring en controle van geneesmiddelen
De Europese Farmacopee biedt handvatten voor het ontwikkelen van een product, maar ook voor het samenstellen van een eisenpakket waaraan het eindproduct moet voldoen. De Farmacopee stelt
gevalideerde analytische methoden, referentiestandaarden en aanvaardbare limieten beschikbaar. Zowel de grondstofproducenten als de eindproductfabrikanten en de registratiehouders maken gebruik van deze informatie bij het ontwikkelen van een product en het opstellen van een registratiedossier. Vervolgens baseren de registratieautoriteiten, de inspectie en andere overheidsinstanties en controlelaboratoria zich ook op de beschikbare standaarden bij het beoordelen en toetsen van farmaceutische producten.
2.3.5.1 Ontwikkeling: stimulering generieke industrie
De generieke industrie heeft voordeel van het bestaan van de Europese Farmacopee doordat men zich kan baseren op alom aanvaarde regels bij het ontwikkelen van geneesmiddelen. De chemische eigenschappen van werkzame bestanddelen worden beschreven in de grondstofmonografie. Ook voor de kwaliteit van veel hulpstoffen wordt verwezen naar de Europese Farmacopee. Vervolgens worden ook voor producten kwaliteitseisen vastgelegd. Dit totaalplaatje aan regels bespaart de fabrikant ontwikkelingskosten en bevordert de uitwisselbaarheid van het generiek met het originele product. Het vereenvoudigde ontwikkelingsproces faciliteert daarnaast de snelheid waarmee generieke producten op de markt kunnen komen. Het registratieproces kan bovendien soepeler verlopen wanneer het een Ph. Eur.-grondstof betreft, zeker wanneer men gebruikmaakt van het certificatieschema (zie
paragraaf 2.3.3.2). Dit alles past in het overheidsstreven om, waar mogelijk, kosteneffectieve zorg te realiseren.
2.3.5.2 Registratieprocedures
In Europa worden geneesmiddelen hetzij op nationaal niveau, hetzij op Europees niveau beoordeeld voordat een handelsvergunning verleend kan worden. De beoordeling van de kwaliteit van een geneesmiddel voor humaan of veterinair gebruik, vindt plaats op basis van onder meer de Europese Farmacopee en verordeningen, richtlijnen en richtsnoeren van de Europese Commissie/EMEA. De beschikbaarheid van een grote verscheidenheid aan gevalideerde analytische methoden, referentiestandaarden en aanvaardbare limieten in de Europese Farmacopee vergemakkelijkt het registratieproces. Bij beoordeling wordt bijvoorbeeld gecontroleerd of de verwijzing naar een
farmacopeemethode correct plaatsvindt, maar de validatie van de betreffende methode hoeft niet meer in twijfel getrokken te worden. Het hanteren van Ph. Eur.-testmethoden en -eisen scheelt zo tijd voor zowel overheid als industrie en beïnvloedt de tijd die nodig is om een handelsvergunning te verlenen of te verkrijgen. Op deze manier wordt de beschikbaarheid van geneesmiddelen bevorderd.
2.3.5.3 Partijgewijze vrijgifte vaccins en bloedproducten
Vaccins en plasmaproducten worden in veel landen partijgewijs vrijgegeven door de overheid. In Nederland vindt de vrijgifte van humane vaccins en (houdbare) bloedproducten plaats bij het RIVM. Deze vrijgifteprocedure betekent dat naast een handelsvergunning ook een additionele
overheidskeuring nodig is. De resultaten van een dergelijke keuring gelden voor de hele EU (mits het product er een handelsvergunning heeft). De kwaliteitseisen van deze biologische producten zijn als productmonografieën in de Europese Farmacopee opgenomen, die deze onafhankelijke keuringen faciliteert.
Het feit dat er een monografie beschikbaar is, betekent dat er duidelijkheid bestaat over de eisen en dat er methoden beschikbaar zijn voor de analyses. Dit alles verhoogt de efficiëntie waarmee vaccins beschikbaar gemaakt worden voor de markt.
2.3.5.4 Inspectie
De overheid controleert de kwaliteit van de geneesmiddelen zowel vóór als ná registratie. Tijdens het registratieproces worden de kwaliteitseisen vastgelegd waaraan de grondstoffen, de productieprocessen en het eindproduct moeten voldoen. De grondstofleveranciers en de eindproductfabrikant dienen te verklaren dat er gewerkt wordt volgens Good Manufacturing Practice (GMP). Deze verklaring is gekoppeld aan wetgeving op het gebied van algemene werkwijzen in de farmaceutische industrie. Of deze verklaring overeenkomt met de werkelijkheid wordt in Nederland getoetst door de Inspectie voor de Gezondheidszorg (IGZ).
Inspecties kunnen zowel vóór als ná registratie plaatsvinden en zijn daarom niet altijd gekoppeld aan een registratiedossier. De Europese Farmacopee vergemakkelijkt de taak van de inspecteurs door het beschikbaar maken van wettelijk geldende methoden. Uitgangspunt voor de inspectie is, waar mogelijk, de controle op uitvoering van de methoden en eisen van de Europese Farmacopee. Omdat daarmee gericht gecontroleerd kan worden, kan de benodigde (kostbare) inspectietijd worden verkort.