Expression profiling of molecular
chaperones in the murine Duchenne
muscular dystrophy model mdx in early
disease stages.
Gwenny Cosemans
Student number: 01808604
Supervisor(s): Dr. Boel De Paepe
A dissertation submitted to Ghent University in partial fulfilment of the requirements for the degree of Master of Science in the Biomedical Sciences
1. Preface
First, I would like to thank Dr. Boel De Paepe for her excellent guidance throughout past two years. I could not have got a better supervisor for a master thesis. I had been given the chance to perform research at a whole new level. I have grown a lot, learned to think more critically and work more independently. In addition, I am grateful that my work could contribute to Caroline Merckx her PhD. She is a motivated, promising researcher, who I admire a lot. She also performed the grip tests and surgically collected muscle biopsies for my work. Thank you for all your help, even with the slightest question, I could come to you. Next, I would like to thank Sophie D'hose for sharing her lab experience with me. All the (few) people in the lab made it so much fun to conduct research and motivated me throughout these two years.
I should not forget about my fellow student colleagues, especially Milena and Elien. The movie nights, dinner parties and boardgame nights where my favorite occasions to relax. You always listened to me and supported me without doubt. I am so grateful that I got to know you two. Elien, hopefully we can continue to perform research (for out PhD’s) and co-house together since the Coronavirus is restricting our search for a new apartment. Truthfully, I could not have achieved my current level of success without a strong support group. First of all, I would like to thank my parents, who supported me with love and
understanding. Secondly, my boyfriend, who made these stressful times more bearable and was always there to cheer me up. Thank you all for the unwavering support.
Hopefully, you are all safe and sound.
2. Preamble
Due to protective measures during the covid-19 pandemic, I was no longer able to work in the lab from March 24th onwards. Luckily, I had, by then, finished most of my practical work. I performed protein extractions in the first year and during summer and conducted western blot analysis and immunofluorescence experiments mainly during the first semester of my second year. However, I could not get a second look at all my immunofluorescence stainings, which is quite essential in discussing my results. Overall, the crisis did not affect my work substantially, and I am glad I was able to finish early, so I could focus more on the written part of my Master Thesis. During the lockdown, I completed my laboratory notebook as requested, and applied some additional statistical analysis to my results. My supervisor and I continued to work together via e-mail, by giving feedback and performing an online conference meeting to practice my presentation.
Table of contents
1. Preface ... i 2. Preamble ... i 3. List of abbreviations ... iv 4. Summary ... 1 5. Introduction ... 15.1 Duchenne muscular dystrophy ... 1
5.2 Pathophysiology... 3
5.3 The dystrophin protein ... 5
5.4 The dystrophin glycoprotein complex ... 6
5.5 Animal models of DMD ... 7
5.6 Heat shock proteins ... 9
5.7 Heat shock factor 1 ...12
6. Materials and methods ...15
6.1 Animals ...15
6.2 Grip strength ...15
6.3 Muscle sampling ...15
6.4 Western blotting (SDS-Page) ...16
6.5 Comparing Image lab (Biorad) with Studio Image Lite (Licor) ...17
6.6 Immunofluorescence ...17
6.7 Statistical analysis ...18
6.8 Student participation in experimental work ...18
7. Results ...20
7.1 Quantitative analysis of HSP70/90 expression via western blot ...20
7.2 Qualitative analysis of HSP70/90 expression via immunofluorescence ...24
8. Discussion ...27
8.1 A need for novel (molecular) therapies for DMD...27
8.1.1 Gene replacement therapy ...27
8.1.2 Exon skipping ...28
8.1.3 Read-through therapy ...28
8.1.4 Nuclease-based gene editing ...29
8.1.5 Dystrophin-independent gene therapy ...29
8.1.6 Stem cell therapy ...29
8.2 Chaperones are upregulated in DMD ...30
8.3 Chaperones localize to muscle fibers and inflammatory cells ...30
8.3.1 Presence of only sparse amounts of revertant fibers in mdx muscle. ...31
8.3.2 HSP70 co-localized with regenerating fibers in mdx muscle. ...31
8.3.3 HSP70/HSP90 co-localized with macrophage marker F4-80 in mdx mice. ...31
8.4 HSP modulation could affect disease progression of DMD ...31
9. Conclusion ...32
10. References ...33
11. Poster ... vi
3. List of abbreviations
AAV
Adeno-associated virus
ABD
Actin binding domain
ACE
Angiotensin-converting enzyme
AD
Activation domain
ADP
Adenosine diphosphate
Aha-1
Activator of Hsp90 adenosine triphosphatase 1
AON
Antisense oligonucleotide
AR
Aldose reductase
ATP
Adenosine triphosphate
BCLAS
The Belgian Council for Laboratory Animal Science
BMD
Becker muscular dystrophy
Cdc37
Cell division cycle 37
CH
Calponin homology domain
CK
Creatine kinase
CRISPR/Cas9
Clustered Regularly Interspaced Short Palindromic
Repeats/CRISPR-associated nuclease/helicase
CTD
C-terminal domain
DAG-FITC
Donkey anti-goat fluorescein isothiocyanate
DAR-CY3
Donkey anti-rabbit cyanine
DBD
DNA binding domain
DGC
Dystrophin-glycoprotein complex
DKO
Double knock-out
DMD
Duchenne muscular dystrophy
DNA
Deoxyribonucleic acid
EARA
European Animal Research Association
ECG
Electrocardiogram
ECL
Enhanced chemiluminescence
ECM
Extracellular matrix
EDTA
Ehylenediamine tetraacetic acid
ER
Endoplasmic reticulum
F-actin
Filamentous actin
FDA
Food and Drug Administration
F4/80
EGF-like module containing mucin-like hormone
receptor-like 1
GAR-POD
Goat anti-rabbit horseradish peroxidase-conjugated IgG
GC
Glucocorticoids
GRMD
Golden retriever muscular dystrophy
GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
HDAC
Histone deacethylase
Hip
HSP70-interacting protein
Hop
HSP organizing protein
HR
Heptad repeats
HSE
Heat shock element
HSF
Heat shock factor
HSP
Heat shock protein
Ig
Immunoglobulin
IL
Interleukin
JAG1
Jagged 1
LZ
Leucine zipper
MD
Middle domain
MOPS
3-(N-morpholino)propanesulfonic acid
MSC
Mesenchymal stem cell
Myf5
Myogenic factor 5
MyoD
Myoblast determination protein
NBD
Nucleotide binding domain
NCAM
Neuronal cell adhesion molecule
NEF
Nucleotide exchange factor
NFAT5
Nuclear factor of activated T-cells 5
NF-κB
Nuclear factor κB
NGS
Next generation sequencing
NMD
Nonsense-mediated decay
NO
Nitric oxide
(n)NOS
(neuronal) Nitric oxide synthase
NTD
N-terminal domain
OCT
Optimal cutting temperature
ORF
Open reading frame
PAX7
Paired box 7
PBS
Phosphate-buffered saline
PCR
Polymerase chain reaction
PEF
Peak expiration flow
PMO
Phosphorodiamidate morpholino oligomer
PTM
Posttranslational modification
RD
Regulatory domain
RNA
Ribonucleic acid
ROS
Reactive oxygen species
SBD
Substrate binding domain
SC
Satellite cell
SERCA
Sarco-/endoplasmic reticulum calcium adenosine
triphosphatase
SIRT1
Sirtuin 1
SMIT
Sodium myoinositol transporter
TALEN
Transcription activator-like effector nuclease
TauT
Taurine transporter
TNF-α
Tumor necrosis factor α
TRIS
Trisaminomethane
UP
Unfolded protein
UPR
Unfolded protein response
UTRN
Utrophin
ZFN
Zinc finger nuclease
4. Summary
Duchenne muscular dystrophy (DMD) is an X-linked, recessive muscle disorder that
ultimately results in life-threatening cardiac and respiratory complications. Mutations result in the absence of dystrophin, a compound of the dystrophin-glycoprotein complex which
connects the cytoskeleton with the myocyte’s extracellular matrix. A dysfunctional complex introduces membrane tears upon contraction, resulting in an enhanced calcium influx, sodium overload, necrosis and edema. Initial regeneration by satellite cells will not suffice at later disease stages, which is characterized by muscle fibrosis. Conventional treatments, including physiotherapy and corticosteroids, improve the patient’s quality of life, although a cure is still absent. Heat shock proteins (HSPs) regulate proteostasis, by refolding proteins or targeting them for degradation, and participate in nearly every cellular process. Among them, HSP70 and HSP90 are the most extensively studied. For this project, we utilized a DMDmdx mice model to determine the expression (western blot) and localization
(immunofluorescence) of HSP70 and HSP90, to evaluate their potential role in early disease stages (4, 8 and 12 weeks). This study revealed HSP70 upregulation in the active
degeneration/regeneration phase of mdx mice, presumably a compensatory mechanism to aid tissue recovery. In addition, HSP70/90 co-localized with infiltrating macrophages of mdx mice, whereas HSP70 were also associated with regenerating fibers. The former results indicate that both HSPs are players in the early inflammatory response, while the latter points to the role of HSP70 in the early compensation mechanism. We suggest that
HSP-modulation, especially HSP70 upregulation, could be considered beneficial for DMD patients as a supportive treatment.
5. Introduction
5.1 Duchenne muscular dystrophy
Muscular dystrophies are a group of inherited muscle disorders characterized by waves of muscle tissue degeneration and weakness. The most common form within the group is Duchenne muscular dystrophy (DMD), a recessive, X-linked disorder in children that affects approximately 1 in 3500 boys born world-wide2. Mutations or rearrangements in the
dystrophin (DMD) gene result in the absence of dystrophin, a protein necessary to provide muscle strength and stability. A milder form of a muscular dystrophy is Becker muscular dystrophy (BMD), a condition wherein mutations in the DMD gene lead to the production of truncated, yet partially functional dystrophin. As a result, patients usually have a later onset of symptoms and a milder clinical progression. Although no cure exists yet, supportive treatments tend to enhance the patients’ quality of life, however, their clinical application is limited4.
Typically, DMD patients show a variable phenotype. Initial symptoms include muscular weakness and atrophy, hypertrophic calf muscles, clumsiness, scoliosis and difficulties with stair climbing or tiptoeing. The diagnostic Gowers’ sign can identify patients. It describes a patient’s use of hands and arms when going from a squatting to a standing position. This sign manifests in patients since they lack hip and thigh muscle strength. Because dystrophin is also present in the gastro-intestinal system, nervous system and endocrine glands, gastric dilatation, intestinal hypo-motility and mood disorders often appear concomitant with DMD6. Because of the progressive character of the disease, it will manifest at later timepoints as: delayed development of motor skills (talking, walking and sitting), loss of ambulation (around puberty), respiratory impairment, muscular weakness and cardiomyopathy. Usually, patients die because of compromised cardiac or respiratory function due to progressive muscle degeneration7.
The diagnosis is usually made in early childhood (between 2-7 years) after symptoms have been noticed. A subsequent first line of tests will be performed. Usually, dystrophin gene deletion and duplication testing will provide information about the mutation and if it disrupted the open reading frame (ORF) of the dystrophin gene. The most commonly used techniques include multiplex ligation dependent probe amplification, comparative genomic hybridization array and multiplex polymerase chain reaction (PCR). When the first line of tests failed (in 30% of the cases), a second line of tests can be applied, which include genetic sequencing of the patient’s genome, often executed via next generation sequencing (NGS). This method can detect point mutations (nonsense/missense mutations) or small mutations (deletions, duplications or insertions). A muscle biopsy will be obtained from the patient if the results of the secondary tests are inconclusive. This biopsy is then subsequently tested for the
presence of dystrophin via immunohistochemistry (qualitative) or via western blot
(quantitative). The neuromuscular disorder may also be diagnosed based on elevated levels of enzymes in the blood, including creatinine kinase, lactate dehydrogenase or alanine aminotransferase. Elevated levels of these enzymes indicate persistent tissue damage6. Mutations in the DMD gene disrupt the open reading frame and introduces a premature stop codon, rendering the patient incapable of producing functional dystrophin (loss-of-function mutation). Due to this premature stop codon, C-terminal truncated transcripts are very unstable and undergo degradation rapidly. Currently, over 3000 mutations have been associated with the disease, predominantly deletion of 1 or multiple exons (60-70%), point mutations (26%) and exon duplications (10-15%). Several mutational hotspots have been identified, but the most prevalent hotspot is situated between exons 45 and 55, which generally results in the removal of the central portion of the rod domain of dystrophin. In the case of BMD, mutations do not cause a shift in the ORF of the dystrophin protein, resulting in the production of a partially functional, truncated form2.
Current DMD interventions focus on alleviating symptoms in patients. To improve the patient’s quality of life, different strategies can be adopted. In general, physicians recommend physiotherapy in combination with glucocorticoid (GC) treatment (e.g.
deflazacort). They can preserve upper limb and respiratory function, delay loss of ambulation and prevent scoliosis surgery in numerous cases. However, additional interventions are required since the long-term administration of GCs are associated with several side-effects6. Physiotherapy includes management at several levels; rehabilitation, endocrine,
gastrointestinal, nutritional, respiratory, cardiac, orthopedic and osteoporosis. Rehabilitation management focuses on minimizing contractures and deformities of muscles and joints, often via physical, speech, language and occupational therapy. Endocrine management is especially required due to the side effect of GCs and will lead to impaired growth, delayed puberty and adrenal insufficiency. Delayed puberty is the result of hypogonadism and can often be cured via testosterone replacement therapy. The benefits outstand the side effects of the hormonal therapy (acne, body odor and behavioral changes). Adrenal insufficiency is a life-threating condition caused by a hypothalamus-pituitary axis imbalance. In this case, GC treatment must be stopped immediately, and intramuscular hydrocortisone injections should be administered at once. Therefore, it is important to monitor growth of the patient, diagnose hormonal deficiencies and prevent a life-threatening adrenal crisis. Gastrointestinal and nutritional management is required to counteract potential weight gain/loss, nutrient/fluid imbalance, dysphagia and mandibular contracture. The primary cause is again from GC treatment, but can be exacerbated due to the disease itself (loss of ambulation that decreases energy expenditure because of immobility). Again, nutritional monitoring (growth/weight) and promoting the intake of a healthy, balanced diet is essential6,7.
GC therapy increase apatite and subsequent caloric intake, together with sodium and fluid retention. For the gastrointestinal tract, managing constipation, reflux and motility is
essential. Osmotic laxatives and proton pump inhibitors could both alleviate these symptoms, that result from abdominal muscle weakness, dehydration and decreased transit time.
Respiratory management aims to prevent atelectasis, mucus plugging and pneumonia (which could ultimately lead to respiratory failure and cardiac arrythmias). Respiratory function is carefully monitored via spirometry, and assisted coughing/ventilation is applied when needed. It is also recommended to immunize DMD patients yearly for Influenza and Pneumonia. Morbidity and mortality are related to both respiratory and cardiac failure and should be the major focus of attention. Cardiomyopathy, heart failure and rhythm
abnormalities can be accessed via electrocardiogram (ECG) and non-invasive imaging. Interventions include angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers and cardiac transplant (being a last resort treatment). Orthopedic management tries to preserve motor function, spine stability and bone health as long as possible. Osteoporosis is another side effect of GC treatment and can be treated via the intravenous administration of biphosphate6,7.
Besides symptomatic treatments, such as physiotherapy, mechanical ventilation and corticosteroids, molecular experimental treatments are currently under investigation as a supportive therapy for DMD. Recent Food and Drug Administration (FDA) approved drugs for the treatment of DMD include Ataluren and Eteplirsen. These compounds are mutation-specific and can only be used in a select number of patients. Ataluren can be used for boys with a premature stop codon in their DMD gene, whereas Eteplirsen is skipping exon 51 in DMD patients. Administration of Ataluren enables larger amounts of functional dystrophin to be produced. In contrast, exon skipping acts on the pre-mRNA level, with the aim to restore the ORF of dystrophin. As a result, the patient switches from a Duchenne-phenotype into a Becker-like phenotype, in which the reading frame is intact, and a partly functional protein is produced. This method uses tailored antisense oligonucleotides (AONs) carrying
complementary sequences to the targeted pre-mRNA. Both therapies attempt to enhance gene product function, but this action is still insufficient to cure patients. In addition, potential molecular therapies are still in the preliminary phase6.
5.2 Pathophysiology
At early disease stages, myofibers of patients show necrosis and cycles of muscle regeneration-degeneration associated with chronic inflammation. Muscle regeneration
(characterized by centrally nucleated myofibers) is a complex process involving degenerating myofibers, inflammation and activation of neighboring satellite cells (SCs) 8. In healthy
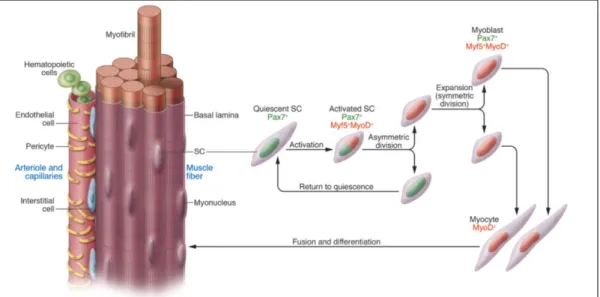
muscle, these muscle stem cells reside in a quiescent state between the sarcolemma of the myofiber and the basal lamina (figure 1) 8,9. Upon disruption of the sarcolemma (i.e. trauma), the persistent intracellular accumulation of calcium activates proteases that cleave various cellular proteins. As a result, an immune response will be generated, recruiting inflammatory cells (neutrophils and macrophages) to the place of injury. These inflammatory cells remove cellular debris and switch the stem cells into an active state 8. Activated SCs undergo asymmetric cell division (figure 1); one fraction proliferates and differentiates, which could either give rise to de novo myofibers or fuse with pre-existing ones. The other fraction generates identical daughter cells, and contributes to the self-replenishment aspect of the stem cell pool 8,9. Quiescent SCs are characterized by paired box 7 (PAX7) expression, whereas activated SCs are positive for PAX7, myogenic factor 5 (Myf5) and myoblast determination protein (MyoD) 9. Premature activation of SCs leads to their exhaustion and impairs myogenesis, which occurs at later disease stages8.
When this compensation mechanism fails, muscle tissue will be progressively replaced with adipose and connective tissue. Damaged myocytes are especially prone to oxidative damage and biomechanical stress, which is prominent in DMD8. A study also revealed that the presence of dystrophin is necessary for the stem cells to polarize and to undergo asymmetric cell division10.
Figure 1 - Asymmetric cell division of satellite cells (SCs) during muscle regeneration.
Muscle stem cells are located between the basal lamina and the sarcolemma of the myofiber. Other myogenic cells, including pericytes and endothelial, hematopoietic and interstitial cells, surround the muscle fiber. Quiescent SCs are characterized by PAX7 expression and replenish the stem cell pool, whereas activated SCs are PAX7+,
Myf5+ and MyoD+ and contribute to newly formed myocytes9.
Although the progression of DMD is unstoppable, the presence of low levels of revertant fibers in patients is quite remarkable. These fibers are able to re-express dystrophin, but they are not abundant enough to compensate for the disease. The splicing machinery of the muscle cell is able to partially bypass the mutation, thereby producing fully functional
dystrophin. This residual dystrophin does not elicit an immune response and has little benefit on muscle function, since it contributes to only 1-7% of all myofibers11. Revertant fibers are now considered as an indicator of muscle activity and serve as an index of muscle
regeneration12.
The absence of dystrophin leads to a dysfunctional dystrophin-glycoprotein complex (DGC) that renders the membrane unstable and increases the susceptibility of the sarcolemma for contraction-induced lesions. An increased membrane permeability allows the abnormal influx and accumulation of calcium within the hypercontracted myocyte 11. Abnormal calcium signaling can be due to membrane-induced tears, Ca2+ influx via stretch-activated or leaky channels and release via ryanodine receptor channels 13. A persistent elevated calcium concentration activates cytosolic proteases that further exacerbate muscle damage by inducing necrosis11. The sarco-/endoplasmic reticulum calcium ATPase (SERCA) pump, normally reducing intracellular calcium levels, bears inactivating modifications which further hamper calcium homeostasis. The impaired buffer capacity of the SERCA pump triggers inflammatory pathways and ultimately muscle degeneration. Inflammation increases secretion and activation of cytokines (pro-inflammatory interleukins IL-1 and IL-6, nuclear factor κB (NF-κB) and tumor necrosis factor α (TNF-α)) and chemokines, thereby improving leukocyte adhesion and complement activation. Inflammation intensifies as the disease progresses. Affected vascular tissue can further aggravate the damage by compromising blood flow to the myocytes11. Another mechanism that contributes to muscle destruction is oxidative stress, which involves the production of reactive oxygen species (ROS) that are retained in muscle cells. DMD is also associated with mitochondrial dysregulation and elevated proteasomal degradation 11,14.
Boel De Paepe et al. demonstrated for the first time the importance of osmolytic pathways in the pathophysiology of DMD. Osmolyte pathways are implicated in normal muscle
functioning and are activated under hypertonic conditions. Hypertonicity leads to water efflux from the cells, which will subsequently shrink. In response, osmolytic protective programs will try to restore normal cell volume and intracellular calcium concentration by accumulating osmolytically active (non-perturbing) organic solutes. These compounds include polyols, such as sorbitol and myo-inositol, and the amino acid taurine. Sorbitol is generated from glucose, which requires aldose reductase (AR) to catalyze the reaction. Taurine and myo-inositol will undergo transporter-mediated uptake, whereby taurine transporter (TauT) and sodium myoinositol transporter (SMIT) utilize the electrochemical concentration gradients of the organic solutes as a driving force 15.
The master regulators of inflammation and osmolytic protection are NF-κB and nuclear factor of activated T-cells 5 (NFAT5) respectively. NFAT5 activation leads to the induction of AR, TauT and SMIT. NF-κB is a transcription factor which induces the expression of tissue-specific adhesion molecules and cytokines, thereby initiating and further amplifying the inflammatory response, which is also prominent in DMD. Ultimately, these factors prevent stress-related cell death. Muscle fibers of DMD patients are characterized by; continuous inflammation with subsequent leaky membranes due to decreased osmotic stability,
increased cytosolic calcium concentration, persistent sodium overload and muscle edema. In response, the body will try to compensate and protect the cells via the activation of NF-κB and NFAT5. As the disease progresses, the compensation mechanisms will fail, and the dysfunctional osmoregulation further contributes to the pathology of the disease. These findings are important evidence in the search for therapeutic targets, since osmolytes have only recently recognized to contribute to the disease 15. However, several papers already suggest the beneficial role of taurine supplementation on the clinical progression of DMD. Long-term administration of this essential amino acid could prevent cardiac dysfunction and improves skeletal muscle function in the mdx mouse model 16,17.
5.3 The dystrophin protein
Dystrophin protects the sarcolemma against contraction-induces mechanical stress. In addition, the protein is involved in calcium homeostasis and nitric oxide (NO) synthesis. It is part of the dystrophin-glycoprotein complex (DGC). Structurally, dystrophin is 79 exons long (2200 kb) and contains 4 functional domains (figure 2); an N-terminal domain, a central rod, a cysteine-rich domain and a C-terminal domain. The N-terminal domain is also called ABD1, which stands for actin binding amino terminal domain 1. ABD1 contains 2 homology domains (CH1 and CH2) and is known as the portion of the protein directly binding to filamentous actin (F-actin), thereby providing a link between the sarcolemma and the cytoskeleton of the cell. The rod domain contains 24 spectrin repeats and a second actin-binding domain (ABD2), providing a second position for actin to bind. Apart from actin, the rod domain can also interact with other microstructures, including microtubules and phospholipids 2. Dystrophin can undergo posttranscriptional modifications (PTM) via phosphorylation of threonine, tyrosine and serine residues that lie in the cysteine-rich region of the molecule, downstream of the spectrin repeats. PTMs are required for the proper interaction of the dystrophin protein with other partners of the DGC 18.
5.4 The dystrophin glycoprotein complex
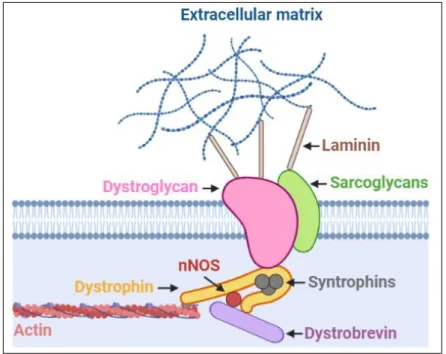
Dystrophin is part of the large dystrophin glycoprotein complex (DGC) which interconnects the cytoskeleton of the muscle cells with its surrounding extracellular matrix (ECM). It serves as a mechanotransducer by translating physical signals from the ECM to changes in gene expression. The DGC stabilizes the plasma membrane of striated muscle cells, and loss of one or several components of the complex leads to plasma membrane instability and myofiber loss. Functionally, the complex can be divided into 2 parts; a structural part, where it protects the sarcolemma from mechanical damage, and a signaling part, where it functions as a scaffold for signaling proteins. The complex can be roughly divided into 3 domains (figure 3); an extracellular one, which contains heavily glycosylated α-dystroglycan, a transmembrane portion, which encompasses β-dystroglycan, sarcoglycans and sarcospan (not shown in figure 3), and a cytoplasmic part, which includes dystrophin, dystrobrevin, syntropins and neuronal nitric oxide synthase (nNOS). Both dystroglycan isoforms are translated from a single transcript but are processed differently. A-dystroglycan acts as a receptor and binds laminin, thereby connecting the muscle cell to the basal lamina. Β-dystroglycan interacts with α-Β-dystroglycan, sarcoglycan and dystrophin, forming a bridge between the extra- and intracellular portion of the complex. As mentioned, dystrophin binds directly to the intracellular actin network. Neuronal NOS (nNOS) is loosely associated with the complex and is activated during exercise; it produces NO, which acts on local blood vessels, dilating them, thereby increasing the blood flow to provide sufficient amounts of oxygen and nutrients to the contracting muscle fibers2.
Figure 2 - Structure of the dystrophin protein.
Dystrophin contains 4 major functional domains. The N-terminal actin-binding domain 1 (ABD1) contains 2 calponin-homology (CH) motifs. The central rod domain harbors 24 spectrin-like repeats (R1-R24, shown in red) with 4 interfering proline-rich hinges (H1-H4, shown in green). Within the central rod domain, a second actin-binding domain (ABD2) is present, spanning R11-R17. The cysteine rich domain and C-terminal domain form the last 2 essential regions in the dystrophin protein 2,3.
Figure 3 - Structure of the dystrophin glycoprotein complex (DGC).
The DGC can be divided into 3 domains. The extracellular domain contains α-dystroglycan, a protein which connects the DGC with the extracellular matrix via laminin. The transmembrane part comprises β-dystroglycan, sargoglycans and sarcospan (not shown here) and provides additional support to the complex. The intracellular portion contains dystrophin, which is the central linker of the complex and binds to actin, syntrophins, dystrobrevin and neuronal NOS. The latter contributes to signal transduction and mechanoprotection 2,4.
5.5 Animal models of DMD
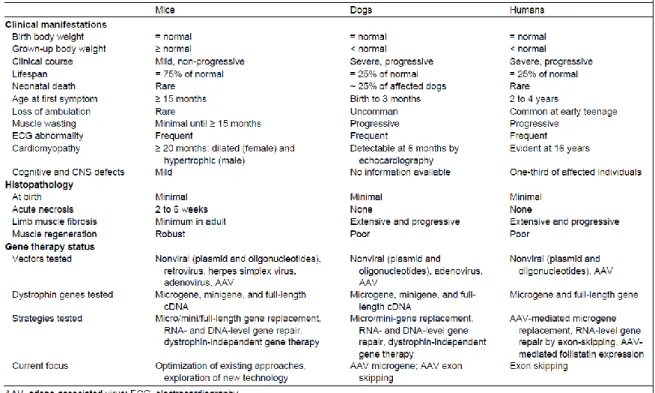
Pre-clinical animal models allow researchers to understand the fundamental processes that underly diseases and help them reveal new potential therapeutic targets. Although different DMD animal models have been established so far, the murine mdx model is predominantly used. The choice of your animal model depends on the research that you want to conduct, and every model has its own advantages and disadvantages. Mouse and golden retriever models are considered the golden standard for studying gene therapy. However, murine models lack the human immune response and data interpretation could be impaired due to their small muscle size compared to human muscle 1. On the other hand, they have a short generation time, are easy to genetically manipulate and are cost-effective 4. Table 1
compares disease severity and gene therapy status in mice, dogs and humans 1. Other animal models include: Caenorhabditis Elegans, Drosophila Melanogaster, Danio rerio and the currently upcoming rat (generated via CRISPR-CAS) and pig models 1,11.
Zebrafish models are useful for screening and evaluating the efficacy and toxicity of low molecular weight drugs. In addition, they could also be used to visualize molecular processes
in vivo, since their larvae are translucent. Homogenous sap mutants contain a nonsense
mutation in the N-terminal domain of dystrophin, which leads to protein loss with subsequent muscle degeneration, inflammation and progressive fibrosis. The major downside of the model is its low translational potential, since zebrafish are non-mammalian4.
The Golden Retriever Muscular Dystrophy (GRMD) model contains a spontaneous mutation in intron 6, which leads to the introduction of a premature stop codon in exon 8. Due to large differences in dystrophic phenotype between individual dogs, data interpretation can be inaccurate 4. A study revealed that the upregulation of the jagged 1 (JAG1) gene is associated with a milder phenotype in GRMD dogs, which could be an explanation for the phenotypic diversity 19. Additional ethical and financial issues further complicate the use of this model 4.
Upcoming pig models are utterly promising because they resemble to humans to a greater extent in terms of anatomy, physiology and genetics. However, they are not used in preclinical studies yet. Transgenic pig models contain a mutation in exon 52 (mutational hotspot) of the DMD gene. Their representation of the human DMD condition is also similar; they show increased creatinine kinase (CK) levels, lack functional dystrophin and fibrosis occurs progressively. Although their resemblance is remarkable, they show upregulation of utrophin, which does not occur in human patients, and results in a milder, BMD-like
phenotype 4. Utrophin, which is encoded by the UTRN gene, shares 80% homology with dystrophin and shows functional redundancy. The protein is abundantly found in the lung, kidney, liver, brain and spleen and in lower levels in skeletal muscle and the heart. Utrophin is especially prominent in utero during skeletal muscle development and will be progressively replaced by dystrophin after birth at the level of the sarcolemma11.
At the moment, the most commonly used murine model for DMD is the spontaneous DMDmdx
mutant mouse on a C57BL/10 background. These mutant mice do not express dystrophin, as in human patients. The nonsense point mutation induces a C-to-T transition in exon 23 of the dystrophin gene, thereby introducing a pre-mature stop codon in this exon1. Colonies are maintained by sibling mating of homozygous males with hemizygous females. Muscle biopsies from these mutants are histologically normal in the first 2 weeks after birth, but abnormalities arise due to muscle weakness, which can be microscopically visualized (fields of necrosis) around 3 weeks. Biochemical analysis reveals elevated serum CK levels, which indicate myopathy, a characteristic of DMD. Subsequent muscle regeneration and limb hypertrophy (except for the diaphragm) act as a compensation mechanism, which tends to be more successful in mice compared to humans. Muscle wasting, heart failure
(cardiomyopathy) and scoliosis occur only in mice older than 15 months. In addition, they also show elevated intracellular calcium levels, persistent activation of NF-κB, chronic
inflammation characterized by the presence of infiltrating macrophages and revertant fibers 4. Noticeable, utrophin is upregulated in regenerating fibers of the mdx mouse, and together with the revertant fibers, decrease the severity of fibrosis and loss of function in myocytes. Utrophin upregulation is not seen in humans, and some questions remain about the accuracy of the model. Despite their low cost and easy accessibility, they show a milder phenotype and near-normal lifespan in contrast to humans affected by DMD, which led to the
development of variants of the mdx mouse model. For example, incorporating exercise in the daily routine of a mdx mouse could drastically increase stretch-induced tears that
exacerbates muscle necrosis and allows more calcium to accumulate in the myocytes. This gives the model a more pronounced phenotype that resembles the human condition better 11. A double knock-out (dko) mdx mouse model shows a more severe phenotype, since both dystrophin and utrophin are absent. These mice are usually smaller in size. The challenge with this particular strain is that they often die prematurely, are difficult to breed and are not representable for the human condition. The development of a heterogenous strain could possibly overcome these challenges. Another approach focuses on reducing muscle
regeneration in mdx mice by eliminating MyoD, a muscle-specific transcription factor involved in lineage commitment (asymmetric cell division) of SCs1. Also, high dose irradiation blocks muscle regeneration and creates a revertant fiber knockout model 4.
The Mdx model on a C57BL/6 background can additionally be treated with chemical mutagens that results in the production of a wide variety of dystrophin isoforms. Therefore, this model is mostly used in comparative studies, especially to study dystrophin function and expression 4.
Mdx52 mice contain a deletion in exon 52 which abolishes full-length dystrophin completely. These mice exhibit muscle necrosis, regeneration and hypertrophy. But most importantly, they lack the expression of 2 other dystrophin isoforms (Dp260 and Dp140) and express lower levels of revertant fibers 4.
DMD null mice are produced via Cre-Lox recombination, which removes the whole DMD gene. As a result, mutant mice are unable to express any dystrophin isoform. In addition, alternative splicing cannot occur, leading to the absence of revertant fibers. This model is particularly useful in transgenic studies, to study the function of the different dystrophin isoforms 4.
5.6 Heat shock proteins
Chaperones play a central role in normal muscle functioning and respond to extensive exercise and cellular stress. They exert both intra- and extracellular functions 8. A molecular chaperone is defined as any protein that will interact with, stabilizes or helps another protein to acquire its functionally active conformation without being present in its final structure20. Proteins are produced on ribosomes in linear chains of amino acids. To obtain functionality, they must fold in a proper 3D conformation. Constant surveillance of the proteome is required for proteostasis, since proteins are structurally dynamic. Folding relies on the cooperation of many weak non-covalent interactions. In addition, this folding process is rate limited by translation and prone to errors, due to the large amount of different structures these linear chains can adopt. Misfolded proteins expose hydrophobic amino acid residues, and their accumulation can be toxic 20. Molecular chaperones include heat shock proteins (HSPs) and can be further classified according to their molecular weight 8. HSPs contribute to proteostasis and provide a balance between protein synthesis, folding, unfolding and turnover. They form a quality control system in all cellular compartments, but the most important checkpoint is at the level of the endoplasmic reticulum (ER), which is an important site for protein production 5. HSPs can refold or target proteins for degradation (proteasomal degradation, autophagy or lysosomal degradation)20.
Misfolded proteins can accumulate upon stress, for example, when heat, irradiation or toxins are exposed to the cell, and will subsequently initiate an unfolded protein response (UPR), which ultimately results in cell death. HSPs are dynamic in behavior and can be adenosine triphosphate (ATP) dependent or independent. They mostly interact with aggregation-prone proteins, non-native polypeptides and proteins which are targeted for degradation 5. Since HSP70 and 90 are the most widely studied HSPs and are the 2 major players involved in protein folding, they will be the focus of our research. They are present throughout many subcellular compartments, including mitochondria, the nucleus, the sarcoplasm and the sarcoplasmatic reticulum 8. Over time, basal HSP70 and HSP90 levels increase with age, an event which which can be correlated to an increased level of oxidative stress 21.They interact with each other via an HSP organizing protein (Hop), which facilitates client transfer. Both can interact with other partners and cofactors, and they rarely work alone 20. In addition, their functionality can be influenced by PTMs in terms of acethylation and phosphorylation 5.
The HSP70 family allow translocation of client proteins across organelle membranes but also guide substrates for refolding, unfolding, disaggregation or degradation 20. They stabilize the misfolded protein in an ATP-dependent way by acting locally on unfolded regions of the substrate 5,20. Two members of the HSP70 family persist in our body; a constitutive HSP73, which is always present in low levels, and HSP72, which is highly inducible upon stress. In case of stress or trauma, HSP72 are secreted into the extracellular environment where it can bind to inflammatory cells (neutrophils and macrophages), thereby contributing to their activation and chemotaxis, as well as the early inflammatory response 8.
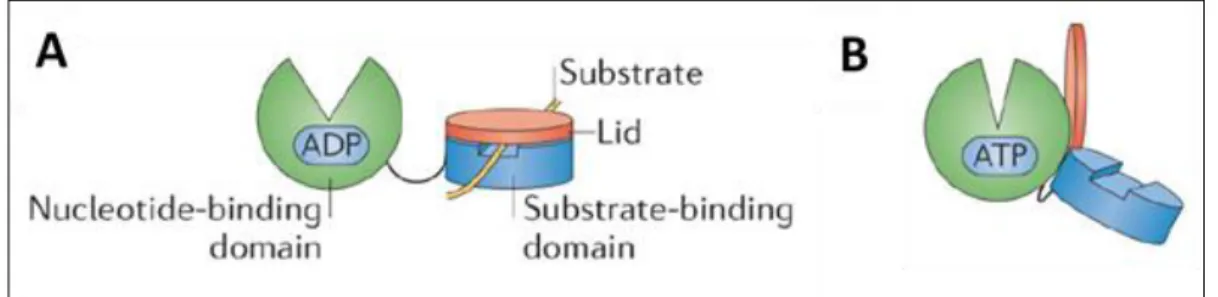
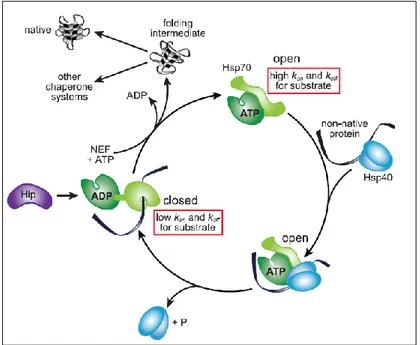
The active conformation of HSP70 persists as an allosteric monomer containing 2 functional domains; an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD). The 2 domains are interconnected via a flexible hydrophobic linker (figure 4)5,20. The NBD contains additionally 4 subdomains, which form a deep cleft upon nucleotide binding. The β-sheet of the SBD serves as the substrate binding site, with an α-helix as a ‘lid’ (highly mobile) to control the access to the site. Figure 5 displays the conformational cycle of HSP70. In the adenosine diphosphate (ADP) bound state, the NBD and SBD are tethered together via the linker, and the lid will cover the substrate binding site (closed conformation). When ATP is bound, the lid will be displaced, away from the β-sheet, promoting client binding (open conformation). Client binding will evoke conformational changes in the HSP itself, thereby exposing regulatory surfaces, allowing them to cooperate with other proteins, which can subsequently modify HSP activity 5,20. These HSP modifiers include nucleotide exchange factors (NEFs), which stimulate ADP release and nucleotide exchange after ATP hydrolysis. In addition, they can also cooperate with HSP40, which stimulate the ATPase activity of the chaperone. HSP40 are capable to accelerate hydrolysis in terms of a 1000-fold, ultimately closing the helical lid, trapping the bound client protein5.
Figure 4 - ADP- and ATP-bound state of HSP70.
The N-terminal nucleotide-binding domain (NBD) and C-terminal substrate-binding domain (CBD) are interconnected via a flexible linker. (A) In the ADP-bound state, the lid covers the substrate bound to the SBD, thereby preventing substrate dissociation. (B) In the ATP-bound state, the lid will be displaced, allowing a new substrate to bind to the SBD. Figure modified from 5.
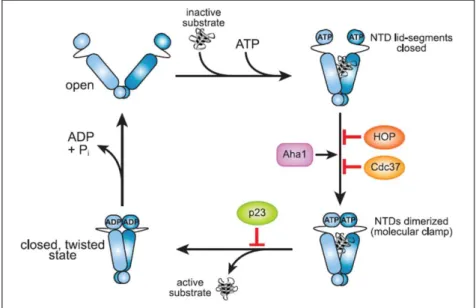
ATP-dependent HSP90 regulate mainly protein trafficking, receptor maturation and signal transduction. Typical client proteins include p53, steroid hormone receptors and kinases. Therefore, they are important players in cell-cycle progression, vesicle-mediated transport and innate immunity. The chaperones will form a homodimer, with each protomer containing 3 distinct regions; an N-terminal nucleotide-binding domain (NTD), a middle domain (MD) and a C-terminal domain (CTD) (figure 6). The NTD binds ATP and participates, together with the MD, in ATP-hydrolysis. The CTD contains a dimerization region. Unique for human HSP90 is the presence of a flexible linker between the NTD and MD, which regulates co-chaperone binding and induces a conformational change. Three distinct conformations can be observed for HSP90, depending on the nucleotide that they are bound to (figure 7). The open Apo-conformation allows binding of ATP and the substrate to the chaperone, which will induce NTD dimerization and subsequent formation of an ATP-hydrolysis site. Regions of the NTD and MD, which contain a similar promotor, will interact with each other, promote ATP-hydrolysis and convert the conformation into a closed ADP-bound state. A final AMP-bound state will readily turn back into the Apo-conformation, ready to bind to ATP and to start the chaperone cycle again5,20.
Figure 5 - Chaperone cycle of HSP70.
HSP40 form a complex with the non-native protein and binds to the ATP-bound state of HSP70 (open conformation). This interaction induces ATP hydrolysis and converts HSP70 into an ADP-bound, closed conformation. Nucleotide exchange factor (NEF) triggers ADP release and rebinding of ATP induces substrate dissociation, making the substrate available for downstream chaperones. HSP70 directly fold protein
intermediates back to their native conformation or pass them down to other chaperone systems. HSP70-interacting protein (Hip) stabilizes the ADP-bound closed conformation, delaying substrate release [19].
Figure 6 – Structure of HSP90.
Functional HSP90 consist of a homodimer, and each monomer contains 3 distinct domains; an N-terminal nucleotide-binding domain (NTD), a middle domain (MD) and a C-terminal domain (CTD)5.
As previously described, ATP-dependent HSP70 and 90 remodeling of client proteins
contributes to protein homeostasis. These dynamic proteins are highly abundant in the under normal physiological conditions, and their expression is further increased in response to stress. HSP70 and HSP90 activity contributes to protein reactivation and requires
additionally co-chaperones, ATP-hydrolysis and client binding by both HSPs. Although both chaperones act in a similar fashion, their substrate recognition is quite different. HSP70 chaperones recognize short hydrophobic or aromatic sequences, which are normally present in the hydrophobic core of folded proteins. In unfolded proteins, these sequences are
exposed, thereby promoting HSP-substrate interaction. In contrast, HSP90 chaperones recognize hydrophobic charged residues over a large surface area, which are also exposed in disordered or unfolded proteins. Interestingly, HSPs possess a holdase activity that can prevent aggregation of bound client proteins. Holdase activity doesn’t require ATP nor co-chaperone binding 5,20.
5.7 Heat shock factor 1
In skeletal muscle, HSPs play additional roles in the modulation of cell signaling, immune response, myogenesis, myofibril assembly and adaptation to physical activity. Stress-induced HSPs are classified as proteins that contain a heat shock element (HSE) in their promotor, which form the target for heat shock factor 1 (HSF1). HSE are composed of alternating inverted repeats containing the sequence ‘nGAAn’ with n representing any nucleotide. The arginine residue of the HSF will interact with the guanine residue of the repeat sequence on the HSE. Under normal conditions, HSF1 is phosphorylated and kept inactive in the cytoplasm. Basal phosphorylation is required to achieve full potency once activated. Stressors induce hyperphosphorylation and nuclear translocation, allowing it to interact with HSE, ultimately resulting in elevated HSP production. Thus, HSF1 protects cells from proteotoxicity and cell death by regulating the expression of HSPs at the transcriptional level. HSFs themselves are regulated by non-coding ribonucleic acids (RNAs), splicing isoforms, changes in oligomerization or compartmentalization and stability 22.
Figure 7 - Chaperone cycle of HSP90.
The open Apo-conformation promotes ATP and substrate binding. Subsequent NTD dimerization traps the bound substrate and creates an ATP-hydrolysis site. ATP-hydrolysis releases in turn the substrate and converts the complex into a closed, twisted conformation. Co-factors Hop and cell division cycle 37 (Cdc37) delay ATP hydrolysis, whereas activator of Hsp90 adenosine triphosphatase (Aha-1) accelerates it. P23 delays substrate dissociation by stabilizing the dimerized structure. ADP release is the final step in this chaperone cycle, and the complex returns to an open state [19].
Six HSF variants are encoded in the human genome, where HSF1 plays the most important role in terms of HSP-dependent protein homeostasis. The general structure of HSF1 (figure 8) is divided into 5 different domains; a DNA binding domain (DBD), a leucine zipper
oligomerization domain (LZ1-3) that contains 2 heptad repeats (HR-A and HR-B), a regulatory domain (RD), a fourth leucine zipper repeat with an HR-C and an activation domain (AD). LZ1-3 are hydrophobic and contain charged residues that form intermolecular leucine zippers when they are aligned upon oligomerization. Deletion of LZ1-3 results in monomeric HSF-1. The RD can undergo PTMs which will affect the stability and activity of the molecule. LZ4 interacts with LZ1-3 and represses oligomerization via hydrophobic and ionic interactions22.
Under basal conditions, the factor remains inactive (monomeric). In the presence of heavy metals, oxidants or toxic agents (stress condition), oligomerization occurs, and the factor assembles into a homotrimer. However, oligomerization alone is insufficient to induce transcription. Therefore, the trimer will translocate to the nucleus via a potent nuclear localization signal. For HSF1, which acts as a thermosensor, unfolding of HR-C and stabilization of HR-A is required for its activation. PTMs on HSF1 include acethylation, phosphorylation and sumoylation, which all will influence the activity and stability of the molecule. Acethylation is a reversible PTM, performed on the lysine residues of the factor via histone acethyltransferase. Eventually, the addition of acethylgroups will promote its stability by preventing proteasomal degradation. In addition, the modified residues decrease the affinity of active HSF-1 for binding deoxyribonucleic acid (DNA). Sirtuin 1 (SIRT1), a histone deacethylase, counteracts this process by removing the acethylgroups. Sumoylation is often seen on lysine residues and on the outer domains of the molecule22.
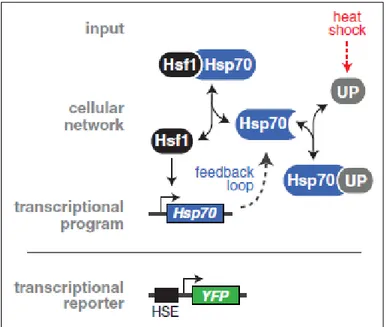
In addition, an ON/OFF chaperone switch influences the action of the transcription factor in a so-called titration model (figure 9). In an OFF-mode, HSF1 is bound to a chaperone and kept inactive, not able to oligomerize. In the ON-mode, HSPs are overwhelmed with unfolded proteins and HSF1 is not their high-affinity substrate anymore. Instead, they bind to unfolded proteins, trying to restore their native structure. As a result, HSF1 is titrated away, and
migrates to the nucleus where it can exert its action. HSP expression will be upregulated and serves as a negative feedback loop, again binding HSF1 when the concentration of unfolded proteins diminishes 23. Research has been contraindicative regarding the major repressor of HSF1. Several studies indicated that HSP70 are the major players acting on HSF1, whereas HSP90 only loosely interact with this factor23,24. Other research suggests that HSP90 are the major repressors and function as a negative feedback mechanism 25,26.
Figure 8 - Structure of heat shock factor 1 (HSF1).
HSF1 contains 5 general domains; a DNA-binding domain (DBD), Leucine zipper 1-3 (LZ 1-3), a regulatory domain (RD), leucine zipper 4 (LZ 4) and an activation domain (AD). The leucine zippers contain 3 heptad repeats (HR-A, HR-B and HR-C). The regulatory domain undergoes post-transcriptional modifications on P-sites that affect the stability and functionality of the molecule [21].
In DMD, both protein families (HSP70 and 90) are enriched in challenged fibers. This points to the initial compensation mechanism as an attempt to restore and regenerate. On the other hand, HSP90 are upregulated in cytotoxic T-cells and macrophages, which are abundantly present in the invading infiltrates at the level of the muscle fiber 27. Researchers are now evaluating whether molecular chaperones could be used as disease markers to identify new treatment options. In addition, targeting chaperone imbalance could lead to a new generation of therapies for protein misfolding disorders, including Alzheimer’s Disease, Huntington’s Disease, Prion’s Disease and Parkinson’s disease 5. However, this type of treatment could only be applied within a restricted time window (so-called ‘window of opportunity’) 28. For example, Parkinson’s disease is characterized by the presence of aggregation proteins, α-synuclein, in so-called neurotoxic Lewy bodies. Interestingly, HSP70 and HSP90 are also present in Lewy bodies, acting as a coping mechanism by limiting the aggregation of α-synuclein. Unfortunately, this mechanism fails rapidly as the disease progresses 29,30. One option to ameliorate the α-synuclein toxicity could be through the overexpression of HSP70. In addition, chemical compounds targeting HSF1 could induce the same effect indirectly. HSF1 modulators could promote nuclear translocation of the transcription factor and upregulation of HSF1 target genes. Also, HSP90 inhibitors could be of possible use. Ultimately, this would lead to the ectopic expression of HSP70 28. Manipulating HSP expression under stress conditions could protect cells, including skeletal muscle. For this project, comparing the expression profile of HSP70/90 in dystrophic and normal muscle could give an indication whether chaperones could be used as a new target in a novel generation of supplementary treatments for DMD.
Figure 9 - Titration model of Heat shock factor 1 (HSF1).
Under basal conditions (OFF-mode), HSF1 is kept inactive in the cytosol via their interaction with HSP70. Upon stress (heat shock), unfolded proteins (UPs) accumulate (ON-mode). HSP70 chaperones have a higher affinity for UPs, thereby dissociating from HSF1. As a result, HSF1 can migrate to the nucleus, where it can bind to the heat shock element (HSE) of its target genes, including HSP70 itself. Over time, HSP70 will be enriched in challenged cells and more available to bind to HSF1 again, serving as a feedback loop [22].
6. Materials and methods
6.1 Animals
All animal experiments were conducted in accordance with the guidelines of The Belgian Council for Laboratory Animal Science (BCLAS/ASBL) and European Animal Research Association (EARA) and were approved by the Animal Ethics committee at the University of Ghent (project numbers ECD18/98 and ECD19/77). Mice were obtained from The Jackson Laboratory, Bar Harbor, Maine. All experiments were conducted on dystrophic mdx
(C57BL/10ScSn-Dmdmdx/J) and non-dystrophic control mice (C57BL/10ScSnJ). Mice were maintained at the University of Ghent on a 12h (reversed) light/dark cycle, under standard condition and with free access to food and drinking water. 16 mice (8 mdx and 8 controls) were sacrificed at 4, 8 and 12 weeks each.
6.2 Grip strength
Before the animals were sacrificed, a four-limb hanging wire test was performed. A plastic hollow box was used (35 cm in height). Soft bedding (a minimum of 3 cm in height) was placed at the bottom of the box to avoid damage to the muscles and to discourage them to intentionally fall from the metal grid. The mouse was placed on the metal grid, turned upside down together with the wire on top of the plastic box, and measured how long it was capable to hang on the grid. For each mouse, 5 tests were performed, and with a time interval of 10 min between each test. The first 2 results were not included in the official data set but were performed to accustom the mouse to its new environment and the test itself. When t = 0, it was recommended to exclude the mouse from the study (drop-out).
6.3 Muscle sampling
Sixteen mice were subjected to cervical dislocation at 4, 8 and 12 weeks. Biopsies were taken from different hind limb muscle groups (m. tibialis anterior, m. soleus, diaphragm, m. gastrocnemius and m. extensor digitorum longus) both in the mdx and wild-type model. For this project, only the m. tibialis was used. For western blot analysis, the tissue samples were immediately placed on dry ice and stored at -80°C. Manipulation was reduced to a minimum and samples were remained frozen until further use in order to obtain robust data and to minimize muscle degradation. It is also possible to snap freeze tissue samples for western blot experiments but was not applied because the former method was more convenient for the direct sorting of samples. The latter method could further limit protein degradation and preserve PTMs, while simultaneously reducing myocyte contaminants and blood that could interfere with downstream processes. For immunological analysis, the snap freezing process was carried out with isopentane cooled by liquid nitrogen. Isopentane reduces the presence of freezing artifacts. An aluminum can was cut in half, filled with isopentane and put in liquid nitrogen until cooled. Dissected tissue was wrapped in aluminum foil and placed in the cooled isopentane for 30 seconds. The samples were transported on dry ice and stored at -80°C until ready for cutting. Tissue was embedded in optimal cutting temperature (OCT) compound. This compound was again used to mount the sample to the chuck of the cryostat. Muscle samples were cut in 8μ thick sections and were mounted on poly-L-lysine coated slides. Slides can be stored for 6-12 months at -80°C until they are fixated with acetone and subsequently stained.
6.4 Western blotting (SDS-Page)
The buffer solution consists of 50mM trisaminomethane (tris) buffer, 5mM ethylenediamine tetraacetic acid (EDTA) and was kept at a pH of 7.4. For each 10ml extraction buffer, 1 tablet of complete protease inhibitor was added. The biopsy was transferred to a petri dish and grinded together with an appropriate volume of extraction buffer (depending on the size of the biopsy). Next, the sample was centrifuged for 15 min at 13 000 rpm at 4°C. Supernatans was pipetted in aliquots of 50 μl each. The protein concentration of each sample was
determined via the Biodrop A280 method. The aliquots were put in a freezer at -80°C.
For each Western blot procedure, the sample with the lowest concentration was taken as the reference. All other samples were diluted according to the reference sample with extraction buffer for equal loading. 12μl of loading buffer and 5μl of reducing buffer was added to each sample. The aliquots were boiled for 2 minutes and cooled down on ice. Subsequently, the buffers for electrophoresis were prepared. The outer buffer consists of 50ml
3-(N-morpholino) propanesulfonic acid (MOPS) running buffer (20x) and 950ml water. The inner buffer consists of 200ml outer buffer and 500μl antioxidant. Samples were loaded onto 10% bis-tris gel (20μl for the samples, 10μl for the molecular standard) and run for 50 minutes at 200V and 120mA. Beforehand, the transfer buffer was made with 849ml water, 100ml methanol, 1 ml antioxidant and 50ml transfer buffer x20. Six to ten pads were soaked in transfer buffer and a sandwich was made (filter-gel-membrane-filter). The sandwich was put in the blot module in the middle of the soaked pads. Proteins were then transferred to nitrocellulose membranes by wet blotting for 60 minutes at 25V and 160mA (NuPage blot). The membrane was washed afterwards with water and tris-buffered saline (TBS). When the membrane was not directly used for the western blot, it was preserved (for maximal 1 week) in TBS and put in the fridge at 4°C.
The membrane was rinsed with water/TBS, and a blocking buffer was made from 1g milk powder and 50ml TBS. The membrane was blocked for 1 hour with blocking buffer. A TBST solution is made and consists of 200 ml TBS and 200 μl tween. The blocked membrane was washed with TBST. Subsequently, the membrane was incubated with the appropriate
antibodies (table 3) overnight and put on a shaker for 2 hours. Enhanced chemiluminescence (ECL) detection of GAPDH (glyceraldehyde 3-phosphate dehydrogenase) and anti-HSP70/HSP90, combined with 1 μg/ml secondary GAR-POD antibody (goat anti-rabbit horseradish peroxidase-conjugated IgG antibody) was visualized via Clarity Max (Biorad). Genetools software (Syngene) was used to compare the expression levels between samples by using GAPDH as the loading control. All solutions and materials for the gel
electrophoresis and transfer were obtained from ThermoFisher.
Protein bands were visualized using the Chemidoc (Biorad). HSP70 and 90 were observed according to their molecular weight, 65-80 kDa and 81-99 kDa respectively. GAPDH has a molecular weight of 37 kDa. Densities of protein bands were calculated with Genetools software (Syngene). The relative raw volume (HSP/GAPDH) was calculated for each track.
6.5 Comparing Image lab (Biorad) with Studio Image Lite (Licor)
To analyze the obtained western blot images, 2 analysis programs were compared with each other to determine the most suitable one. Image lab analyses the volume (i.e. light intensity) of the 3D peak (i.e. lane width x band width). Adjusting color/contrast settings did not affect the densitometric values. The boundaries were automatically detected and manually
adjusted. The program allows the researcher to easy adjust for background noise which can be corrected for each band. After adjusting the boundaries and correcting for the background noise, a data excel sheet was imported that contains several parameters which are further explained in table 2.
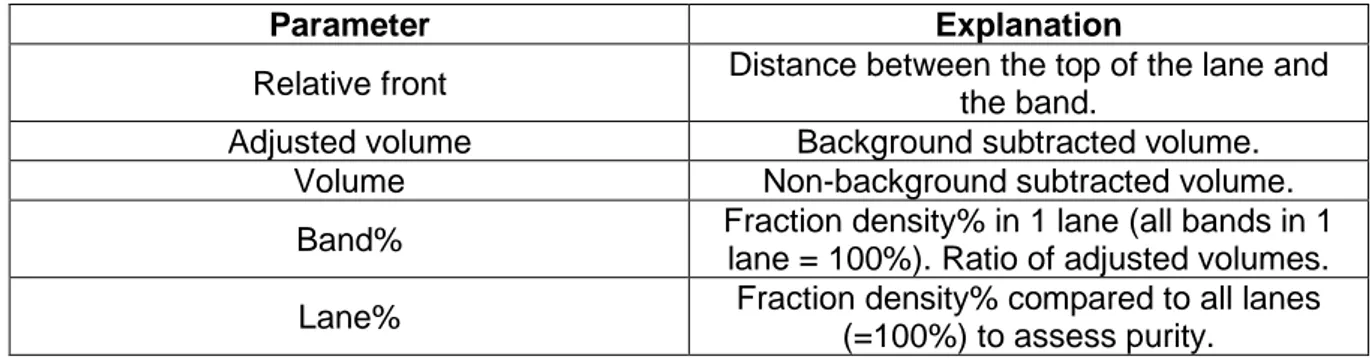
Table 2 – List of parameters encountered by using Image Lab (Biorad)
Parameter Explanation
Relative front Distance between the top of the lane and the band.
Adjusted volume Background subtracted volume. Volume Non-background subtracted volume. Band% Fraction density% in 1 lane (all bands in 1
lane = 100%). Ratio of adjusted volumes. Lane% Fraction density% compared to all lanes
(=100%) to assess purity.
The Licor software, although free of use, was not optimal to process the western blot images. After the image was imported, the program automatically inverted the data and color of the image. The problem was the source of the image, coming from a third party. By solving this problem, one had to make a screenshot of the image of interest (.jpg format), paste it in an empty Word-document and alter its color/contrast settings. Still, the imported image in Studio Image Lite was supersaturated, leading to distorted results. In addition, changing the rotation of the image was also interfering with the values, as was not the case for Image Lab.
Furthermore, altering background subtraction greatly affected the results. Therefore, the Image Lab software form Biorad was used to visualize the western blot images, as being the most reliable/accurate one.
6.6 Immunofluorescence
Histological 8μ sections were prepared from different muscle biopsies, placed on a cover slip and fixed in acetone (stored at 4°C) for 2 minutes. They were subsequently blocked with a phosphate-buffered saline (PBS, Humane+/Donkey+) solution for 30 minutes. During the blocking period, the different antibody solutions were made (appropriate concentrations are made via dilution with PBS). Every used antibody is listed in table 3. A double
immunofluorescence staining was performed to detect both the antigen and cell type qualitatively. Primary and secondary antibody combinations were derived from different species, in order to prevent cross-reaction. The samples were incubated with 100 μl of the primary antibody solutions A and B for 30 minutes each and washed afterwards with PBS. Next, the samples were incubated with the appropriate secondary antibody solution
containing 4 μl/ml donkey anti-goatfluorescein isothiocyanate (DAG-FITC, green) or donkey anti-rabbit Alexa 488 Alexa488, green) and 4 μl/ml donkey anti-rabbit cyanine (DAR-CY3, red) in 1000 μl, and 100 μl solution was added to each sample. After the washing step with PBS, the samples were fluoromounted and visualized under a fluorescence microscope.
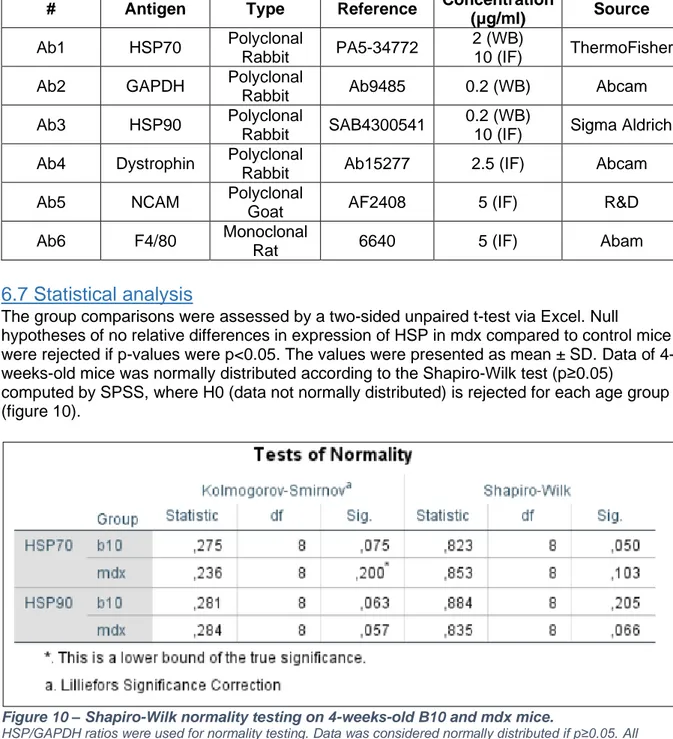
Table 3 - Antibodies used for western blot and immunofluorescence.
6.7 Statistical analysis
The group comparisons were assessed by a two-sided unpaired t-test via Excel. Null
hypotheses of no relative differences in expression of HSP in mdx compared to control mice were rejected if p-values were p<0.05. The values were presented as mean ± SD. Data of 4-weeks-old mice was normally distributed according to the Shapiro-Wilk test (p≥0.05)
computed by SPSS, where H0 (data not normally distributed) is rejected for each age group (figure 10).
Figure 10 – Shapiro-Wilk normality testing on 4-weeks-old B10 and mdx mice.
HSP/GAPDH ratios were used for normality testing. Data was considered normally distributed if p≥0.05. All groups were not statistically not significant. Abbreviations; df = degrees of freedom, sig = significance.
6.8 Student participation in experimental work
I started my internship in November 2018 by learning how to perform protein extractions and western blot analysis (detection of HSP70 and GAPDH). Afterwards, I executed the same experiments on already isolated m. tibialis biopsies and processed protein extracts of 86-weeks-old mice. In December, dr. Boel De Paepe and me performed a double
immunofluorescence staining of SLC5A3 and Pax3/7 on m. tibialis slides of 86-week-old B10 and mdx mice (n=4). The Western Breeze procedure was the last experiment I learned in 2018. For this experiment, the same blot from previous western blot analysis was used to detect GAPDH. In February, the stained slides were analyzed with a fluorescence
microscope (ZEISS) and additional software (Olympus) to evaluate the efficacy of the different antibodies. These antibodies were not included in my project, as they served to practice my skills.
# Antigen Type Reference Concentration
(μg/ml) Source
Ab1 HSP70 Polyclonal
Rabbit PA5-34772
2 (WB)
10 (IF) ThermoFisher Ab2 GAPDH Polyclonal
Rabbit Ab9485 0.2 (WB) Abcam Ab3 HSP90 Polyclonal
Rabbit SAB4300541
0.2 (WB)
10 (IF) Sigma Aldrich Ab4 Dystrophin Polyclonal
Rabbit Ab15277 2.5 (IF) Abcam Ab5 NCAM Polyclonal
Goat AF2408 5 (IF) R&D Ab6 F4/80 Monoclonal
From March 2019 onwards, I started with my own project. Caroline Merckx trained me for the grip strength test and surgical isolation of muscle biopsies (diaphragm, m. tibialis, m. soleus, m. gastrocnemius and m. extensor digitorum longus). After a few observations, I was able to isolate the muscle biopsies myself. For the strength grip test, we experimented with the height of the bedding material, the box and the metal grid where the mice were placed on. Afterwards, I was allowed to perform the experiment on my own. Every experiment related to animal handling and surgery was executed under her supervision. After the behavioral testing and weighing, mice were euthanized via cervical dislocation, which she performed herself. Next, she taught me how to prepare histological sections from the preserved muscle biopsies via a cryostat (Microm 550). Caroline mounted the biopsies on a cork via OCT compound. I accustomed to the cryostat and practiced with different muscle types
(diaphragm, m. tibialis, m. gastrocnemius, m. extensor digitorum longus and m. soleus) and tried to produce ±16 slices/ muscle biopsy.
In September, I spent most of my time in the neuropathology lab processing muscle samples into protein extracts, which we subsequently stored in aliquots of 50μl. The number of
aliquots produced/muscle sample depended on its protein concentration after centrifugation. In total, 24 B10 and 25 mdx m. tibialis biopsies of 4-, 8- and 12-weeks-old mice were
processed. In some cases, the protein concentration was too low, and the amount of solution extracted was insufficient to include the muscle biopsy in the western blot analysis. In total, I prepared 99 B10 and 86 mdx aliquots containing protein extract. Next, I calculated how much buffer should be added to each sample, in order to correct the variable protein
concentrations. Before the actual western blot analysis was conducted, me and dr. Boel De Paepe compared 3 methods: (i) ECL detection of HSP70, HSP90 and GAPDH, (ii) ECL detection of GAPDH, western breeze detection of HSP70 and HSP90 and (iii) western breeze detection of GAPDH, ECL detection of HSP70 and HSP90. Method (i) showed the least background staining and I performed this method on all my western blot experiments. To refresh my memories, dr. Boel De Paepe performed the HSP70/GAPDH western blot analysis of 4-weeks-old B10 and MDX mice that I included in my project (2 blots/age group). The rest of the western blot experiments were carried out on my own. Western blot analysis of 8-weeks-old B10 and mdx mice were repeated with the remaining protein extracts, due to an improper ECL detection of the antibodies. To analyze the expression levels of HSPs and GAPDH, I first compared Studio Image Lite (Licor) and Image Lab (Biorad) software
programs to detect the most accurate one. I applied the same analysis software on all my ECL images. Subsequently, I conducted an unpaired, two-sided student t-test on my relative ratio’s (HSP70/GAPDH) to determine any statistically significant differences in HSP70 expression in B10 versus mdx mice. I also demonstrated that all (relative) results obtained from the western blot experiments were normally distributed via SPSS.
For the next part of my internship, I performed double immunofluorescence stainings on m. tibialis biopsies of 4, 8 and 12-weeks-old B10 and mdx mice. Before I conducted these experiments, I performed a titration of HSP70 (10, 5 and 3.33 μg/ml) and HSP90 antibodies (40, 20, 10 μg/ml) and evaluated their performance under a fluorescence microscope. Subsequently, I performed a double immunofluorescence staining to detect HSP70. I also executed a double HSP70-CD86 staining, but the latter antibody was not efficient, so dr. Boel De Paepe ordered a new antibody which targeted F4/80, another macrophage marker.