EVALUATION OF THE INVOLVEMENT
OF NOTCH1 VARIANTS IN CAROTID
AND VERTEBRAL ARTERY
DISSECTION
Maarten Pannecoucke
Student number: 01504335Supervisors: Prof. Dr. Olivier Vanakker, Dr. Hemelsoet
A dissertation submitted to Ghent University in partial fulfilment of the requirements for the degree of Master of Medicine
Deze pagina is niet beschikbaar omdat ze persoonsgegevens bevat.
Universiteitsbibliotheek Gent, 2021.
This page is not available because it contains personal information.
Ghent University, Library, 2021.
Table of contents
1. Introduction ... 3
1.1. Spontaneous internal carotid and vertebral artery dissection ... 3
1.1.1. Pathophysiology ... 3 1.1.2. Symptoms ... 4 1.1.2.1. sICAD ... 4 1.1.2.2. sVAD ... 4 1.1.3. Diagnosis ... 5 1.1.4. Treatment ... 6 1.1.5. Molecular genetics ... 6 1.2. NOTCH genes ... 8
1.2.1. Structure of Notch receptors ... 8
1.2.2. Notch signaling pathway ... 9
1.2.3. Notch in the vasculature ... 10
1.2.3.1. Notch in vascular development ... 10
1.2.3.2. Notch in adult arteries ... 11
1.2.4. NOTCH and cardiovascular disease ... 12
1.2.4.1. NOTCH1 in Adams-Oliver syndrome and aortic valve disease ... 12
1.2.4.2. NOTCH3 in CADASIL ... 13
2. Aims and objectives ... 15
3. Design and methods ... 16
3.1. Settings and samples ... 16
3.2. Literature search ... 16
3.3. Genomic DNA isolation ... 17
3.4. Primer design ... 17 3.5. PCR ... 17 3.5.1. Pre-PCR ... 17 3.5.2. Post-PCR ... 18 3.6. Next-generation sequencing (NGS) ... 18 3.6.1. Pooling ... 18 3.6.2. Sample preparation ... 19 3.6.3. Sequencing ... 19 3.7. Sanger sequencing ... 19 3.7.1. PCR clean-up ... 19 3.7.2. Sequencing ... 20 3.7.3. Sanger clean-up ... 21
3.7.4. Evaluation of Sanger sequencing results ... 21
3.8. Evaluation of variants ... 21
4. Results ... 24
4.1. Patient characteristics ... 24
4.2. Identified NOTCH1 variants ... 24
4.2.1. Structural characteristics ... 27
4.2.2. Allele frequencies ... 27
4.2.3. Functional impact ... 28
4.2.4. Conservation of nucleotides and amino acids ... 28
4.2.5. In silico predictions ... 29
4.2.5.1. Missense predictions ... 29
4.2.5.2. Splicing predictions... 30
5. Discussion ... 32
5.1. Exon variants ... 33
5.1.1. Variants located in EGF-like repeats... 33
5.1.2. Variants located in other protein domains ... 36
5.2. Intron variants ... 38
5.3. Implications of these results ... 39
5.4. Recommendations for further research and clinical practice ... 40
5.5. Limitations ... 41
6. Conclusion ... 42
7. References ... 43
8. Supplemental Materials ... i
Supplemental table 1. Overview of cysteine variants causing CADASIL. ... i
Supplemental figure 1. Example of a Fragment Analyzer report ... vi
Supplemental table 2. Primers used in this study ... vii
Supplemental table 3. Patient list ... ix
Supplemental figure 2. SeqPatient reports of confirmed variants ... xvii
1
Samenvatting
AchtergrondSpontane dissecties van de grote arteriën in de hals, meer bepaald de carotis interna en de vertebralis arterie, zijn relatief zeldzaam. Desalniettemin zorgen deze samen voor ongeveer 20% van de cerebrovasculaire accidenten bij patiënten jonger dan 45 jaar. De precieze oorzaken van deze dissecties, die per definitie optreden in afwezigheid van voorafgaand trauma, zijn ongekend. Er wordt vermoed dat genetische factoren een belangrijke rol spelen in deze pathologie. Waarschijnlijk gaat het voornamelijk om genetische varianten die op multifactoriële wijze een dissectie veroorzaken. Daarentegen is het niet uitgesloten dat in sommige gevallen een dergelijke dissectie als een monogenische aandoening zou kunnen beschouwd worden. Dit blijkt uit het feit dat ze kunnen voorkomen in het kader van syndromale aandoeningen. Varianten die leiden tot zulke aandoeningen buiten beschouwing gelaten, zijn er actueel nog geen varianten geïdentificeerd waarvan bewezen is dat ze tot dissecties van de carotis interna of vertebralis arterie leiden. In deze masterproef wordt onderzocht of varianten in het NOTCH1 gen betrokken zijn in de pathogenese van deze dissecties.
Methoden
De studiepopulatie, bestaande uit 49 patiënten die in het UZ Gent gediagnosticeerd werden met (een) spontane dissectie(s) van de carotis interna en/of de vertebralis arterie, werd aan de hand van targeted enrichment en next-generation sequencing gescreend op de aanwezigheid van NOTCH1 varianten. De gevonden varianten werden geëvalueerd op mogelijke pathogeniciteit.
Resultaten
Er werden 42 verschillende NOTCH1 varianten gevonden, waaronder 10 die nog niet eerder gerapporteerd werden. Aan de hand van Sanger sequenering werden 6 van de gevonden varianten bevestigd.. Hiervan werd er 1 beschouwd als een mogelijk monogenische oorzaak van spontane dissecties. Verder werden er 24 varianten beschouwd als potentieel multifactoriële oorzaken.
Besluiten
Deze studie toont aan dat NOTCH1 varianten mogelijks betrokken zijn bij het ontstaan van spontane dissecties van de carotis interna en vertebralis interna. Op basis van de vondst van een variant die leidt tot de substitutie van een extreem geconserveerd cysteïne residu in een welbepaald proteïnedomein van Notch1, wordt er vermoed dat soortgelijke varianten een monogenische vorm van dissecties kunnen veroorzaken. Een analoog mechanisme is reeds bewezen in het NOTCH3 gen voor cerebrale autosomaal dominante arteriopathie met subcorticale infarcten en leukoencefalopathie (CADASIL)
2
Abstract
BackgroundSpontaneous dissections of the major arteries of the neck, more specifically the internal carotid and vertebral artery, are relatively rare. Nevertheless, together they account for around 20% of the cerebrovascular accidents in patients under the age of 45. The exact causes of these dissections, which by definition occur in the absence of preceding trauma, are unknown. It seems likely that genetic factors play an important role in this pathology. The majority of the dissections can probably be explained by genetic variants which cause disease in a multifactorial way. It cannot be excluded, however, that in some cases a dissection can be considered as a monogenic disease. This is supported by the fact that they can occur in the context of syndromic disorders. Apart from variants in such disorders, as of today there are no variants identified which are proven to lead to spontaneous dissection of the internal carotid and vertebral artery. This study investigates if NOTCH1 variants could be involved in the pathogenesis of these dissections.
Methods
The study sample, consisting of 49 patients diagnosed with (a) spontaneous dissection(s) of the internal carotid and/or the vertebral artery at the Ghent University Hospital, was screened for the presence of NOTCH1 variants by targeted enrichment and next-generation sequencing. The identified variants were evaluated for potential pathogenicity.
Results
In total, 42 different NOTCH1 variants were identified, of which 10 were novel variants. Sanger sequencing allowed to confirm 6 variants. Of these variants, 1 was considered as a potential monogenic cause of spontaneous dissections. Furthermore, 24 variants were interpreted as possible multifactorial causes.
Conclusions
This study demonstrates that NOTCH1 variants are potentially involved in the development of spontaneous dissections of the internal carotid and vertebral artery. As a variant leading to the substitution of an extremely conserved cysteine residue in a well-defined protein domain of Notch1 was identified, it is assumed that such variants can cause these dissections in a monogenic manner. A similar mechanism has been described in NOTCH3 for cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).
3
1. Introduction
1.1. Spontaneous internal carotid and vertebral artery dissection
Cervical artery dissections (CAD) can be divided in internal carotid artery and vertebral artery dissections (ICAD & VAD), depending on which one of the two major arteries in the neck is affected (1). Dissections of the internal carotid and vertebral artery can be considered spontaneous when occurring in the absence of craniocervical trauma (2). In traumatic dissections, an external cause is evident. Spontaneous internal carotid and vertebral artery dissection (sICAD & sVAD) are both fairly rare phenomena, as supported by their annual incidence of 2.5 to 3 per 100 000 for sICAD and 1 to 1.5 per 100 000 for sVAD. Together, sICAD and sVAD are responsible for 2% of all ischemic strokes. In young adults however, these dissections account for approximately 20% of all ischemic strokes (3). With sICAD occurring more frequently than its vertebral counterpart, it can be concluded that together they form an important cause of stroke in patients younger than 45 years of age.
1.1.1. Pathophysiology
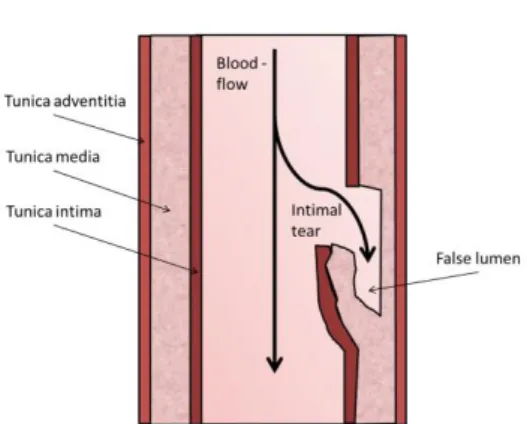
One or multiple tears in the inner layer of the vascular wall, the tunica intima, lead to the collection of blood in the wall of the artery, thereby forming an intramural hematoma. This separates the arterial layers creating a false lumen (3,4) (Figure 1). The extracranial segments of the carotid and vertebral artery are believed to be prone to injury due to their mobility and the proximity of bony structures such as the styloid process (3).
A dissection can lead to an ischemic stroke following two possible mechanisms. The first and most common mechanism is through the activation of platelets and the coagulation cascade, leading to thrombus formation. Eventually, the thrombus travels to the cerebral arteries where it can block the passage of blood, resulting in an ischemic stroke. Alternatively, the intramural hematoma compresses the normal lumen of the artery. The reduced blood flow or even complete occlusion leads to hemodynamic insufficiency and eventually a stroke (4).
Figure 1. Mechanism of an arterial dissection
4 1.1.2. Symptoms
1.1.2.1. sICAD
The manifestations of both sICAD and sVAD can be categorized in local and ischemic symptoms (3). Local symptoms often form the initial presentation of sICAD, more specifically pain usually precedes the other symptoms by 3-4 days (5). Listed according to the frequency of occurrence, the pain can be of the following nature: unilateral headache which frequently develops gradually (2/3 of patients) (5), unilateral facial or orbital pain (1/2 of patients) (5) and unilateral neck pain (1/4 of patients) (5). Partial Horner’s syndrome, which is a combination of miosis and ptosis, is a local symptom with a high specificity (high true positive rate) but a low sensitivity (low true negative rate) for the diagnosis of sICAD, with only half of the patients presenting with this symptom. The presence of this symptom, even in the absence of all other symptoms, suggests the diagnosis of sICAD until proven otherwise. It should be distinguished from complete Horner’s syndrome, in which anhydrosis forms the third symptom (3). Other local symptoms, occurring only in a minority of patients, include cranial nerve palsies and pulsatile tinnitus (3,6). Importantly, 50-95% of sICAD patients suffer cerebral or retinal ischemic symptoms. The most severe symptom in this context, an ischemic stroke, is frequently preceded by transient monocular blindness or transient ischemic attacks. However, 20% of patients have an ischemic stroke without any preceding signs (3).
1.1.2.2. sVAD
As in sICAD, pain is frequently seen as the first manifestation of sVAD. Two possible types of pain are observed. The first, found in around 2/3 of patients (5), is a unilateral headache which is similar in characteristics to the headache described in sICAD. However while sICAD headache is typically located in the frontotemporal area, the headache occurring in patients with sVAD is located in occipital area (1,5). Neck pain forms the second type of pain and is present in almost half of patients with sVAD (5), which is therefore twice as frequent as in sICAD patients (5). It should be noted that both symptoms can be bilateral, although less frequent. These initial symptoms are less distinct than those of sICAD and the diagnosis of a musculoskeletal problem could mistakenly be made. A striking difference between both symptoms is the median interval between onset of pain and occurrence of other symptoms. Which can be simultaneous to 14.5 hours for the headache and 2 weeks for the neck pain (1,5). Other possible symptoms which are present in approximately half of patients include vertigo, unsteadiness, nausea, vomiting and unilateral facial numbness (7). Finally, ischemic symptoms occur in at least 90% of sVAD patients (3). Lateral medullary syndrome or Wallenberg's syndrome, a typical kind of stroke caused by damage to the brain stem, is found in less than 20% of the patients (1,3). The typical presentation of both sICAD and sVAD patients is listed in table 1 (3).
5
Table 1. Typical presentation of a patient with sICAD/sVAD
1.1.3. Diagnosis
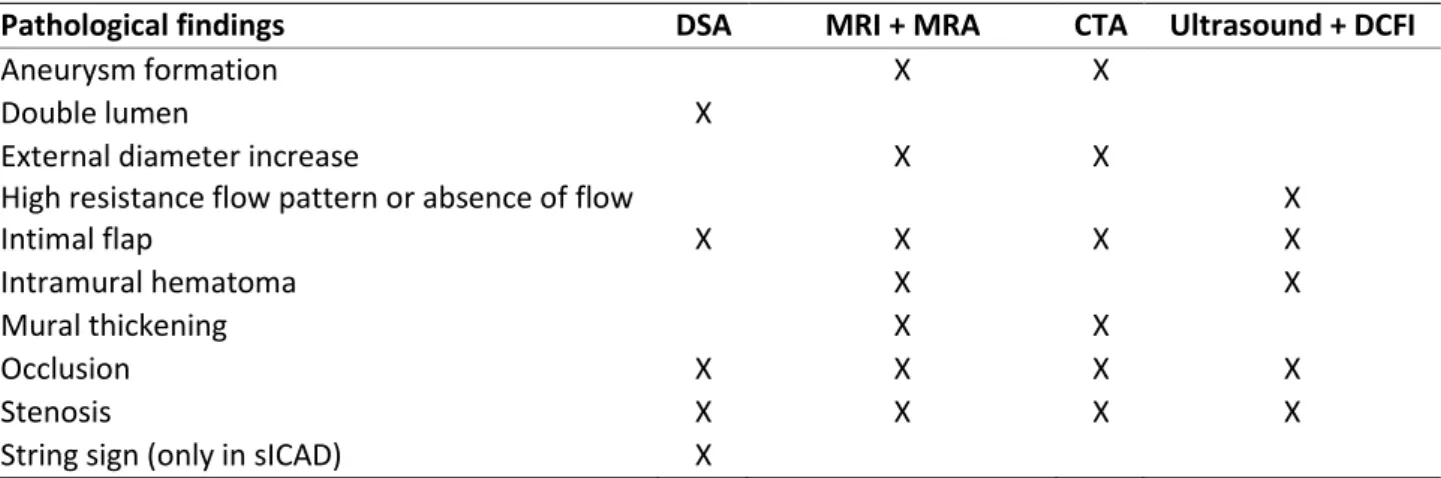
There are four frequently used imaging techniques that can confirm the diagnosis of cervical dissections. These techniques visualize the intramural hematoma and/or the arterial lumen, which can be narrowed or occluded (1). Digital subtraction angiography (DSA) is an invasive imaging modality that used to be the standard in the diagnosis of carotid and vertebral dissections (2,6). In DSA, contrast medium is injected and the image before the injection is subtracted from the image after injection (4). In the last decade, DSA has been replaced by the less invasive magnetic resonance imaging (MRI) and MR angiography (MRA) as standard in diagnosis (3). Because of the vertebral artery’s smaller diameter and varying vessel caliber in the population, the sensitivity and specificity of both techniques is lower for sVAD than for sICAD. Another non-invasive technique which has replaced DSA, used for visualizing both the arterial lumen and wall, is computed tomography angiography (CTA). In patients with a carotid artery dissection an abnormal blood flow pattern can be detected using ultrasonography in combination with doppler color flow imaging (DCFI). This technique is less sufficient for the diagnosis of a vertebral dissection, as the vertebral artery is hard to visualize with ultrasound due to the proximity of bony structures (2). Depending on the chosen technique, different pathological findings characteristic for sICAD and sVAD are detected (1–3,6) (table 2).
sICAD sVAD
Unilateral head, facial or neck pain Headache or pain in the back of the neck Cerebral or retinal ischemia Cerebral ischemia (posterior circulation) Partial Horner’s syndrome
It should be noted that less than one-third of sICAD patients presents with this triad. If a patient presents with two out of three symptoms, the diagnosis should be suspected (3).
Pathological findings DSA MRI + MRA CTA Ultrasound + DCFI
Aneurysm formation X X
Double lumen X
External diameter increase X X
High resistance flow pattern or absence of flow X
Intimal flap X X X X
Intramural hematoma X X
Mural thickening X X
Occlusion X X X X
Stenosis X X X X
String sign (only in sICAD) X
DSA: Digital substraction angiography, MRI: Magnetic resonance imaging, MRA: Magnetic resonance angiography, CTA: Computed tomography angiography, DCFI: Doppler color flow imaging
Table 2. Pathological findings detected by imaging techniques in sICAD/ sVAD patients.
6 1.1.4. Treatment
As sICAD’s and sVAD’s often heal spontaneously, prevention of neurological damage is the main focus of treatment. The aim is to restore blood flow and prevent thromboembolic complications (2,3). The first treatment option is thrombolysis, in which the blood clot is dissolved and blood flow is restored (6). Associated risks include extension of the intramural hematoma, subarachnoid hemorrhage and pseuodoaneurysm formations. However, data suggest that the occurrence of such events is low (8). Antithrombotic therapy, on the other hand, focusses on the thromboembolic mechanism. This treatment can be subdivided in anticoagulation and antiplatelet therapy. Both treatments have specific indications and the choice should be based on the patient’s individual situation. For instance, anticoagulation therapy is opted in patients with severe stenosis or occlusion, while antiplatelet therapy is preferred in patients with a large infarct (6). The placement of stents for treatment of CAD remains controversial, due to its association with thromboembolic complications and occlusion (4). It could be used however in the following situations: persisting symptoms despite medical treatment, the presence of contraindications to antithrombotic therapy, a significant limitation of cerebral blood flow due to stenosis or when an expanding dissecting aneurysm is suspected (1,6). Finally, surgical interventions, such as artery ligation or clipping, are linked to severe complications and should therefore be limited to complex cases (1,6).
1.1.5. Molecular genetics
Ultrastructural connective tissue abnormalities have been frequently detected in patients with spontaneous dissections, suggesting that sICAD and sVAD patients have an underlying structural defect of the arterial wall (9). Indeed, a higher risk for sICAD and sVAD is associated with autosomal dominant polycystic kidney disease (ADPKD) and some inheritable connective tissue disorders (such as Marfan’s syndrome, Ehler-Danlos syndrome type IV, Osteogenesis imperfecta type I), in which vascular abnormalities occur. However, only a small fraction of patients (1-4%) are identified with one of these disorders (10,11). On the other hand, angiography could identify traits of fibromuscular dysplasia in 8-16% of sCAD patients. The prevalence of FMD is higher in sICAD than in sVAD patients (1).
Some potential genetic risk factors have already been identified. For instance, the common single nucleotide polymorphism (SNP), c.665C>T (rs1801133; in literature often referred to as C677T), in the gene coding for methylenetetrahydrofolate reductase (MTHFR) was found to be associated with spontaneous CAD (sCAD) (12,13). The c.665C>T polymorphism causes a substitution of alanine into valine at position 222 (p.Ala222Val) and results in a thermolabile variant of the enzyme, the activity of which is only half of that of the normal enzyme (13). People who have the MTHFR 665TT genotype reportedly have elevated levels of cysteine, as given enzyme finds its role in the conversion of homocysteine (14).
7 An association seems plausible, as hyperhomocysteinemia could damage the arterial wall leading to sCAD (13). So far the exact role of the c.665C>T polymorphism in sICAD and sVAD remains unclear. Multiple association studies on this matter have been conducted, 4 of them found an association between the MTHFR 665TT genotype and CAD (13,15–17). On the other hand, an equal amount of studies failed to demonstrate a significant association between both, thus rejecting the homocysteine-hypothesis (14,18–20). One of these studies did however find a significant association in patients with multiple dissections (14). A meta-analysis, including the majority of the cited studies, showed an overall significant association (21). However, it was stated that this finding should be replicated in a larger, independent sample of CAD patients.
Mutations in the TGFBR1 and TGFBR2 genes, which encode for transforming growth factor β receptors 1 and 2, could form another potential genetic risk factor. However, data on this subject is limited. One study identified two novel disease causing mutations in the TGFBR2 gene of two patients with sCAD, indicating that TGFBR2 mutations may be a cause of these dissections (22). No other studies replicating these findings were found. Results of these studies, substantiating the role of genetic risk factors in sICAD and sVAD, indicate that further research into such risk factors is needed.
A case report found a rare missense variant in the NOTCH1 gene in a 44 years old female suffering from recurrent dissections of the vertebral and carotid arteries. In this patient only hypertension and migraine were present; other known risk factors were ruled out by an extensive set of examinations. Previous traumatic incidents were ruled out, labeling the dissections as spontaneous. Both infectious risk factors and hyperhomocysteinemia were absent in this patient. Connective tissue disorders were ruled out by a dermatologist, and no signs of hypermobility or dermic lesions were present. Furthermore, angiography used to detect fibromuscular dysplasia was negative. No other vascular or cardiac problems were found. However, it should be noted that her family history showed her mother had a thoracic aortic aneurysm 10 years earlier. A comprehensive analysis of genes associated with inherited aortopathy and connective tissue disorders, which was ordered because of the maternal antecedent, revealed a heterozygous variant of unknown significance in exon 34 of the
NOTCH1 gene. The single nucleotide variant c.6365C>T resulted in the substitution of proline
at codon 2122 to leucine (p.Pro2122Leu), of which the prevalence in population databases is low (0.0000082). In silico predictions provided by Sorting Intolerant From Tolerant (SIFT), Polyphen-2 and Mutation Taster unanimously suggested that the variant is likely to be disruptive. The same variant was also found in the patient’s mother, while test results for other family members weren’t mentioned in this case report (23). So far, the potential link between genetic variation of the NOTCH1 gene and spontaneous CAD has not been investigated.
8 1.2. NOTCH genes
The Notch signaling pathway, which is an evolutionarily conserved pathway, finds its main purpose in the regulation of cell fate and cellular functions such as proliferation, differentiation, and survival. It is an intercellular signaling mechanism, mediating the interaction between membrane-bound ligands with receptors on the cell surface of neighboring cells. In mammals, the main actors include four receptors (Notch 1-4) and five ligands (Delta-like 1,3 and 4 and Jagged1 and 2) (24).The Notch1 protein is encoded by the NOTCH1 gene, which is located on chromosome 9. More specifically the gene is located within locus 9q34.3. The RNA transcript of this gene contains 34 exons.
1.2.1. Structure of Notch receptors
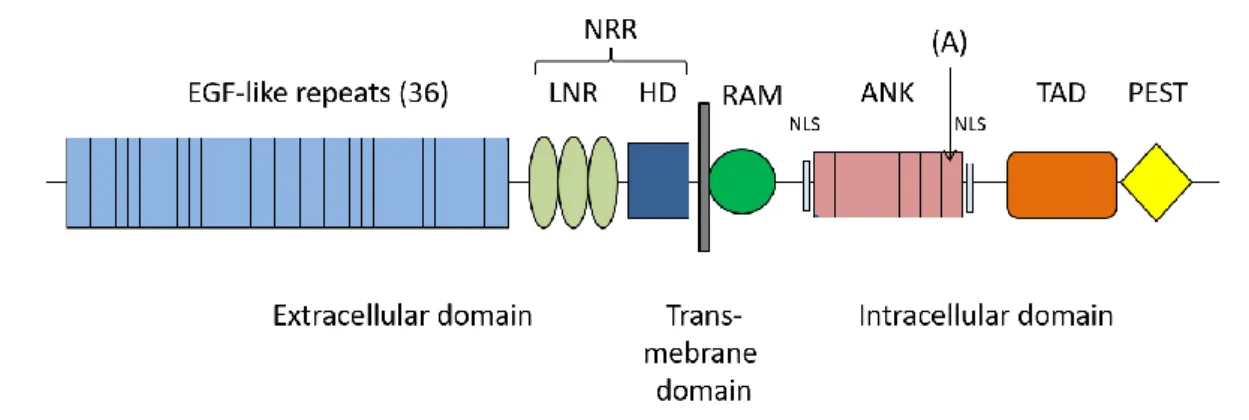
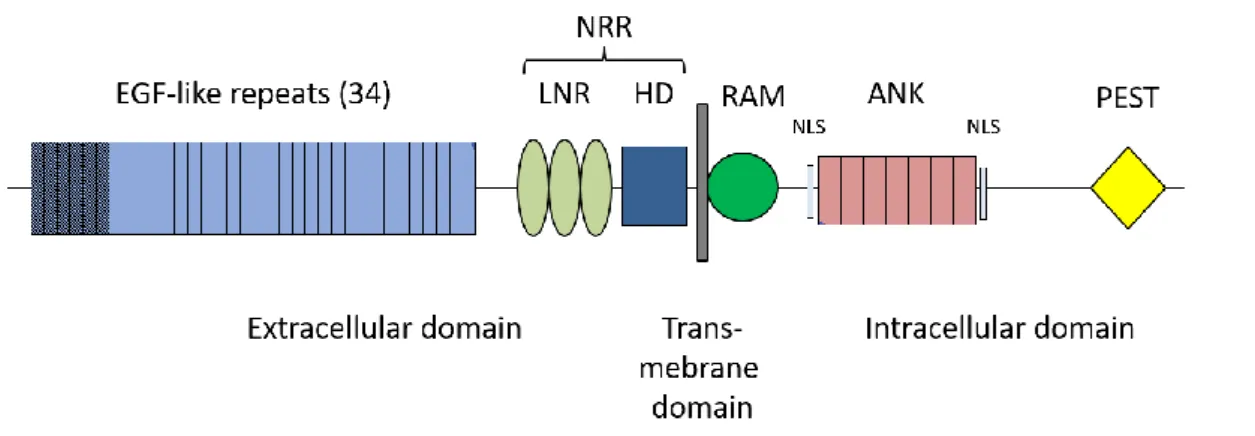
All four Notch receptors are transmembrane proteins and consist of an extracellular domain (NEC), a transmembrane domain and an intracellular domain (NIC) (figure 2) (24,25). The NEC contains multiple tandem repeats of an epidermal growth factor (EGF)-like sequence, the number of repeats varies in the four receptors from 29 (Notch4) to 36 (Notch1). Half of these EGF-like repeats are calcium binding (62). In addition, three cysteine-rich LIN12/Notch repeats lie within the NEC (LNR). The EGF-like repeats are involved in ligand binding, while LNRs inhibit signaling in absence of ligands (26,27).
Together with a heteromerization domain (HD), the LNRs are part of the negative regulatory region (NRR) (28). The NIC domain consists of a recombination signal-binding protein for immunoglobulin kappa J region (RBP-Jκ) associated module (RAM), seven Ankyrin repeats (ANK) flanked by two nuclear localization sequences (NLS), and a transactivation domain (TAD). The latter is only present in Notch 1 and 2. At the C-terminus is a proline glutamic acid serine threonine (PEST) motif (24).
Figure 2. Structure of Notch 1 EGF: Epidermal growth factor, NRR: Negative regulatory region,
LNR: LIN12/Notch repeats, HD: Heterodimerization domain, RAM: RBP-Jk associated module,ANK: Ankyrin repeats, NLS: Nuclear localization sequence, TAD: Transactivation domain, PEST: Proline glutamic acid serine threonine, (A): variant identified in 44y/o with recurrent sICAD (23)
9 The NOTCH1 variant p.Pro2122Leu, found in the aforementioned sICAD patient involves the replacement of the last amino acid of the sixth Ankyrin repeat. Since the Ankyrin repeats mediate protein-protein interactions, a change in this region could potentially affect the protein’s functionality (23).
1.2.2. Notch signaling pathway
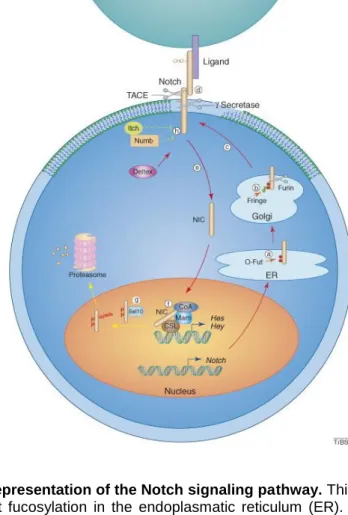
In order to become functional, Notch undergoes several post-translational modifications. A first modification involves the fucosylation by o-Fut in the endoplasmatic reticulum (24,25). In the Golgi network Notch is cleaved by a Furin-like enzyme, which leads to heterodimer formation with both fragments being held together by the HD (24,27,29). Additional glycosylation steps occur as Notch passes through the Golgi system. Finally, processed Notch is exported to the cell membrane, where it can be activated by a ligand (Figure 3) (24).
Figure 3. Schematic representation of the Notch signaling pathway. This illustration was made by
Wu et al. (24). a) O-fut fucosylation in the endoplasmatic reticulum (ER). b) Cleavage in the Golgi network by furin-like enzyme. c) Exportation to the cell membrane. d) Ligand binding, resulting in cleavage by TNF--converting enzyme (TACE) and ϒ-secretase. e) Translocation of the intracellular domain (NIC) to the nucleus. f) Interaction of the NIC with CBF1/SuH/Lag1 (CSL) and mastermind protein (Mam), leading to activation of hairy/enhancer of split. Steps g) and h) are not discussed in this study.
10 In short, a membrane-bound ligand and the Notch receptor interact at the cell surface and initiate a signal that is conducted to the cell nucleus where it regulates the transcription of certain target genes (24,30). In more detail, ligand binding induces conformational changes to the receptor, resulting in the exposure of two sites. The extracellular site is cleaved by a metalloproteinase TACE (TNF--converting enzyme), while the intracellular site is cleaved by ϒ-secretase. This second cleavage releases the NIC, allowing this domain to translocate to the nucleus (24,30,31). At the nucleus, NIC functions as a transcriptional cofactor by binding with the CSL (CBF1/SuH/Lag1) transcription factor (30). In the absence of NIC, CSL is a constitutive repressor of Notch target genes (25). The newly formed complex then interacts with a mastermind (Mam) protein, resulting in the activation of Notch target gene transcription. Next to its role as a co-activator, Mam also induces the degradation of NIC (25,30). The target genes include the hairy/enhancer of split (HES) and HES-related repressor proteins (HERP) family, which act as transcriptional repressors (24,30).
Besides the above discussed canonical pathway, Notch can also interact with components of other signaling pathways, independently of CSL, although the mechanisms of the alternate signaling pathways are not fully understood (32). For instance, Notch can regulate Wnt/β-catenin signaling, which is an important developmental and cellular regulator (33).
1.2.3. Notch in the vasculature
Notch signaling is present in nearly all tissues, with both distinct and overlapping tissue expressions of the four types of receptors. Notch1 is expressed in the brain, heart, vasculature, lung, liver, kidney, intestine, bone marrow, skeletal muscle, thymus, eye and spinal cord (24,34). For this study we will only focus on the importance of Notch in the cardiovascular system, as this system is the most relevant in the discussion of sCAD. Over the last decade, evidence showed that Notch signaling is a key factor in both development and physiology of the vasculature (34). Early studies stressed its importance in vascular morphogenesis, and more recently it became clear that Notch is also required in the adult arteries (35).
1.2.3.1. Notch in vascular development
Vasculogenesis is defined as the formation of a vessel by aggregation of de novo formed endothelial cells. During development, vessels will differentiate into either arteries or veins. It has become clear that the Notch pathway favors the arterial fate of the vessels. In the endothelial cells, Notch stimulates the expression of Ephrin-B2, which is typically found in arteries, but not in veins (32,34). Arterial fate requires the signaling of Notch in a continuous way, as cessation of signaling leads to loss of arterial specification (35).
11 Another important concept in the construction of the vasculature is angiogenesis, i.e. the formation of a vessel from a preexisting vessel (34). More specifically the vascular plexus that is formed by the vasculogenesis is remodeled during angiogenesis (36). The two actors in this process are the tip and stalk cells. Notch is required to promote the specification of these two cells. Particularly Notch promotes the formation of stalk cells and suppresses the tip cell formation (34,35). Endothelial Notch1 is a crucial factor in this process, as pointed out by animal studies. For instance, targeted deletion of endothelial Notch1 was shown to be embryonically lethal in transgenic mice due to cell death and aberrant developmental processes, including angiogenesis (36). In zebrafish (Danio rerio), the interaction between Notch and vascular endothelial growth factor (VEGFA), a key regulator in angiogenesis, was first demonstrated, stressing the importance of Notch signaling for vascular development (37). In both arteriovenous differentiation and angiogenesis, Notch acts downstream of the VEGFA pathway (38).
Finally, the recruitment of mural cells, which differentiate into pericytes or vascular smooth muscle cells (VSMCs), is required to support the endothelial cells. This is the initial phase of vessel maturation. When these mural cells arrive at the vessel, they communicate with the endothelial cells. Endothelial Notch ligands will activate Notch signaling in the mural cells, leading to maturation and arterial differentiation (32,35).
1.2.3.2. Notch in adult arteries
In adult arteries, the Notch pathway is involved in homeostasis and remodeling in the vessel wall. Notch1 and Notch4 are expressed in quiescent endothelium whereas homeostatic VSMCs express Notch3. Communication through Notch signaling between both cell types is needed to maintain homeostasis and to respond to external stimuli (39).
Notch1 is necessary for the inhibition of endothelial cell proliferation, it does so by promoting cell cycle arrest (35). A study reported the role of Notch1 as a mechanosensor in the adult arteries. NOTCH1 is upregulated by shear stress and it ensures flow-dependent endothelial cell quiescence (40).
Furthermore, Notch1 is needed for the regulation of cell-cell junctions. The same study reported that Notch1 promotes the junctional integrity of endothelial cells in stabilized blood vessels (40). Both features suggest that Notch1 ensures the maintenance of the endothelial barrier under the influence of flow forces. Through its role as a mechanosensor, Notch1 is also responsible for promoting and maintaining arterial homeostasis. Endothelial Notch1 is being linked with inflammation, although its exact role remains unclear, and data is still ambiguous (35).
12 The aforementioned study suggests that atheroprotective flow upregulates the expression of endothelial Notch1. In mice, subjected to hypercholesterolemia, loss of endothelial Notch1 led to an increase of atherosclerosis (40).
Next to these endothelial functions, Notch1 also has a role in the regulation of VSMCs. Although Notch1 is absent in VSMCs in homeostatic conditions, it is supposed that after vascular injury, Notch1 mediates VSMC proliferation and neointimal formation. A study suggested the crucial role of Notch1 in VSMC proliferation, migration, and survival, whereas this was first thought to be the domain of Notch3 (41).
1.2.4. NOTCH and cardiovascular disease
1.2.4.1. NOTCH1 in Adams-Oliver syndrome and aortic valve disease
In humans, heterozygous mutations in the NOTCH1 gene are the cause of Adams-Oliver syndrome 5 (AOS5, OMIM # 616028) and aortic valve disease-1 (AOVD1, OMIM # 109730), both developmental disorders with vascular abnormalities (42,43).
Adams-Oliver syndrome is a rare disease characterized by aplasia cutis congenita of the scalp vertex and terminal transverse limb defects. Next to these typical traits, vascular anomalies and congenital heart defects are often present in these patients. Possible vascular anomalies include: pulmonary- and portal hypertension, retinal hypovascularisation and cutis marmorata telangiectatica congenita. These anomalies are more frequent in AOS caused by NOTCH1 mutations in comparison to other causal genes. Five heterozygous NOTCH1 variants leading to AOS are reported in a study analyzing 14 patients within 12 families. The first variant was a de novo deletion of the NOTCH1 5’ region, which includes the promoter partially and the first exon entirely. The second one was a canonical splice-site variant (c.743-1G>T), which affects the exon 5 acceptor splice-site. The three remaining variants were missense variants: c.1285T>C (p.Cys429Arg), c.4487G>A (p.Cys1496Tyr) and c.5965G>A (p.Asp1989Asn). The same study considered NOTCH1 variants as the most common cause of AOS (43).
AOVD, in the context of NOTCH1 mutations, is caused by early developmental defects of the aortic valve and calcium deposition. A study consisting of two families found two different variants leading to AOVD. A nonsense variant (c.3322C>T) leading to the substitution of arginine to a premature stopcodon (p.Arg1108Ter) and a single base pair deletion (position 4515) resulting in a frameshift mutation (p.His1505del) (42). It should be noted that none of these variants, found in AOVD and AOS patients, are located within the same exon as the variant observed in the aforementioned case report.
13 1.2.4.2. NOTCH3 in CADASIL
Another indication for the possible involvement of NOTCH1 variants in sICAD and sVAD, which in turn can lead to stroke, is to be found in one of the other members of the Notch receptor family. Stroke is a typical symptom of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). In particular, CADASIL is characterized by cerebrovascular disease and dementia, with a mid-adult onset for both symptoms. This hereditary small vessel disease is caused by distinct mutations in the NOTCH3 gene (44). The protein structure of Notch3 is largely similar to the earlier discussed structure of Notch1. The number of EGF-like repeats is slightly lower for Notch3 than for Notch1, as Notch3 has only 34 EGF-like repeats compared to the 36 repeats in Notch1. Another difference in the structure of the two proteins, is the absence of the intracellular TAD domain in Notch3 (Figure 4) (24,28). Together with its clear involvement in the vascular system, and its association with AOS5 and AOVD1, this leads to the assumption that NOTCH1 is a candidate as a genetic risk factor for sICAD and sVAD
In CADASIL, causal variants are characterized by a loss or gain of a cysteine residue in the EGF-like repeats of the Notch3 protein (44,45). The EGF-like repeats, which are also present in Notch1, typically contain 6 cysteine residues, with each pair of cysteines resulting in the formation of a disulfide bridge (46). It is stated that alteration of the number of cysteine residues, resulting in an uneven amount, leads to disruption of the formation of disulfide bridges, which in turn leads to disturbance of the secondary structure of Notch 3 (47).
More specifically, the typical pathogenic variants identified in CADASIL patients, are heterozygous NOTCH3 missense mutations which alter the amount of cysteine residues in of the protein’s EGF-like repeats. The majority of these variants is located in the 4th EGF-like repeat (44).
Figure 4. Structure of Notch3 EGF-like repeats 1-6 are shaded, indicating that variants in these
domains cause classical CADASIL, while variants in the non-shaded repeats (7-34) are associated with a milder phenotype (48). EGF: Epidermal growth factor, NRR: Negative regulatory region, LNR: LIN12/Notch repeats, HD: Heterodimerization domain, RAM: RBP-Jk associated module, ANK: Ankyrin repeats, NLS: Nuclear localization sequence, TAD: Transactivation domain, PEST: Proline glutamic acid serine threonine
14 Recently, the position of the pathogenic variant was described as the most important determinant of severity of CADASIL. Variants located in EGF-like repeats 1-6 cause classical CADASIL, while variants located in EGF-like repeat 7-34 are associated with a milder phenotype of the disease (48). In practice this corresponds to a 12-year earlier onset of stroke, lower survival time and greater lesion load as observed by brain MRI for patients carryings the first group of variants (48).
Regarding the type of the CADASIL causing variants, some seem to deviate from the aforementioned characteristics. In addition to the expected heterozygous variants, homozygous and compound heterozygous variants are also found in CADASIL patients. These do not seem to differ from the phenotype seen with heterozygous variants. Furthermore, loss or gain of cysteine residues caused by small deletions, duplications and splice site mutations, instead of the typical missense variants, has been described as a cause of CADASIL (44). Currently, there is a lot of uncertainty regarding the pathogenic role of NOTCH3 missense mutations not involving a cysteine residue. Such variants have been indicated as causes of CADASIL by numerous reports. For most of them, however, this association was not proven as either screening was incomplete or confirmation of the clinical diagnosis by skin biopsy was lacking. Incomplete screening can lead to the typical cysteine altering mutations, present in 1 of the unscreened exons, being overlooked. For the variants p.Arg75Pro, p.Arg213Lys, p.Ala1020Pro and p.Tyr1098Ser, complete screening and confirmation by skin biopsy were performed in the individual studies in which they were reported. Based on segregation analysis and allele frequency (AF), Rutten et al considered p.Arg75pro as the only mutation likely of causing CADASIL. It was stated that given mutation could not be excluded as a cause of CADASIL or a CADASIL-like phenotype (44). A more recent study, which did not take segregation analysis into account, considered 2 new mutations as potentially pathogenic. These variants are p.Arg61Trp and p.Asp80Gly. In addition, p.Arg75Pro and p.Arg213Lys, 2 mutations already reviewed by Rutten et al, were considered potentially pathogenic (49). An overview of all cysteine variants causing CADASIL, as reported by Rutten et al in 2014 (44), is added in the supplemental section (Supplemental table 1).
15
2. Aims and objectives
sICAD and sVAD are both possible life-threatening events, which form an important cause of stroke in young adults. Despite the severity of both conditions, current knowledge about the exact pathogenesis of these dissections is limited. Earlier studies have investigated possible associations between variants in various genes and these types of dissections, yet so far the relationship between the identified genes and sICAD or sVAD is not fully understood. Despite the essential role of Notch1 in the vasculature and its involvement in the pathogenesis of other diseases characterized by vascular disorders, no studies have yet been conducted to look into the possible association between NOTCH1 variants and sCAD. However, one case report recently identified a NOTCH1 mutation as a possible cause of sICAD and sVAD.
Therefore this study aims to further investigate the relationship between NOTCH1 variants and the development of sICAD or sVAD. To do so, NOTCH1 variants will be identified in patients with a known diagnosis of sICAD or sVAD, by means of targeted enrichment of NOTCH1 followed by next-generation sequencing (NGS). Furthermore, each variant will undergo evaluation to determine the mutational spectrum of NOTCH1 and the pathogenicity of each variant
16
3. Design and methods
3.1. Settings and samplesThis study is a descriptive study, more specifically it was conducted according to the design of a case-series. Blood samples were obtained from patients who were diagnosed with unexplained sICAD or sVAD at the Ghent University Hospital (UZ Gent). Patients with known trauma and other known genetic risk factors or causes of stroke or cervical dissections were excluded from this study. Patient samples were pseudonymized as a D-number followed by 7 digits (DXXXXXXX), in a way that only the supervisor can trace back the D-number to the name of the patient. All patients, whose DNA was used in this study, were part of a larger study on stroke, with the aim of identifying causes for their stroke. They all agreed to participate in this study through a written informed consent. This study was approved by the UZ Gent medical ethics committee in accordance with the declaration of Helsinki.
3.2. Literature search
Literature referred to in this study was found through Pubmed. First, literature discussing
NOTCH1 in relation to sICAD and/or sVAD was searched. This was done by entering the
search term “Notch1” in combination with “carotid artery dissection”, “vertebral artery dissection” and “cervical artery dissection”. The first combination led to the case report mentioned earlier. No other results were found through these combinations. Further literature was then sought separately for NOTCH1 and sICAD/sVAD. The snowball method was used, as references mentioned in the case reports were used to find articles on both subjects. Additional literature on dissections of the carotid and vertebral artery was found by using following search terms: “spontaneous”, “carotid artery, internal” “vertebral artery” “cervical arteries” “dissection” “imaging” “treatment” “symptoms” “gene” “MTHFR” and “TGFβR”
Articles about the basics of NOTCH1 and its functions in the vasculature were found with the terms “notch1”, “notch1 receptor”, “notch signaling pathway”, “vasculature”, “arterial”, “vascular development” and “homeostasis”. Articles handling on NOTCH1 in the context of specific diseases irrelevant for this study were excluded. Furthermore, literature discussing CADASIL and vascular diseases associated with NOTCH1 mutations was found through the reference list of a review discussing the role of Notch receptors in human cardiovascular disease. The reference list was constructed through the Mendeley reference management software, in this program the Vancouver style of referencing was selected.
17 3.3. Genomic DNA isolation
Genomic DNA (gDNA) of the patients was present in the laboratory and was previously isolated from blood samples by either the ReliaPrepTM Large Volume HT gDNA Isolation System kit (Promega) or the QIAamp DNA Mini Kit (QIAGen). The gDNA concentration and quality was measured with Trinean dropsense 96. gDNA was diluted with double distilled and ionized water (Sigma-Aldrich) to working solutions of 20 ng/µL.
3.4. Primer design
Primers were designed to meet the requirements of the diagnostic workflow at the Center for Medical Genetics Ghent (CMGG), Belgium. For this, the DNA sequence of the NOTCH1 gene was identified using the Ensembl GRCh37 genome browser. Primer pairs were designed to amplify exons of NOTCH1 using the Primer3 primer design tool (25,26) and are listed in the supplemental materials section (Supplemental table 1). Finally, the specificity of the primer pairs was validated using BiSearch and UCSC in silico PCR. The latter software program was also used to check if there were any single nucleotide polymorphisms (SNPs) or other known variants present in the primer pair. The primers were ordered from Integrated DNA Technologies (IDT) and diluted in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) to 100 µM for stock solutions and 2 µM for working solutions. The primers used during Sanger sequencing are called universal M13-primers, as these can bind to the M13-tail attached to the target specific primers used in PCR reactions. The M13-tail has a universal sequence, which is ‘tgtaaaacgacggccagt’ for the forward primer and ‘caggaaacagctatgacc’ for the reverse primer. 3.5. PCR
3.5.1. Pre-PCR
To avoid contamination, PCR reactions were prepared in a designated work space, the pre-PCR laboratory, which is physically separated from anything that has been in contact with post-PCR products. Components for one post-PCR reaction are listed in table 3. First, master mixes for each primer pair were prepared, after which 7.5 µL of mastermix was added to each DNA sample in a 96-well microtiter plate.
Table 3. Components for one PCR reaction
Component Volume per reaction
Kapa 2G Robust Hotstart ReadyMix (Kapa Biosystems) 5 µl
Primer pair (2µM in TE-buffer) 2.5 µl
18 3.5.2. Post-PCR
The microtiter plate was transferred to a 2720 thermal cycler (Applied Biosystems) in the post-PCR laboratory. The cycling conditions used are described in table 4.
The 96-capillary Fragment Analyzer (Advanced Analytical Technologies) was used to evaluate the PCR product, based on the length (quality) and concentration (quantity) of the fragments. The Fragment Analyzer is a microchip-electrophoresis system in which DNA fragments are separated from each other by high voltage migration trough a conductive gel matrix. This matrix is formed by the capillaries of the chips, which are filled with the gel. The position of each fragment in the matrix is measured through its fluorescence signal. Based on the fact that large fragments are characterized by slower migration trough the gel than the smaller fragments, an accurate estimation of the length of each fragment can be made. The length of these fragments is expressed by the number of base pairs (bp) of which they consist. Furthermore the concentration of the fragments with the same length is measured. Fragment Analyzer reports were consulted to check the results (supplemental figure 1).
The DNF-915 dsDNA Reagent kit (Advanced Analytical Technologies), compatible for the separation of fragments ranging from 35 to 5.000 bp, was used. For this, 2 µL of PCR product was added to 22µl of TE buffer per sample and further handled by the responsible lab technicians. Further handling steps included: gel preparation, buffer preparation, plate preparation, running the plate and uploading the results. The quality of the PCR-product was considered sufficient when one peak, with the expected base pair size, could be observed. 3.6. Next-generation sequencing (NGS)
3.6.1. Pooling
After validation of PCR products, patient-specific pools for the NOTCH1 gene were made. This was done by pooling all NOTCH1 amplicons, 2µL per amplicon, together for each patient. Pooled samples were stored at -20°C until further processed by the lab technicians responsible for operating the MiSeq Personal Sequencer. This procedure consists of sample preparation followed by sequencing.
Table 4. Cycling conditions for PCR
Step Time Temperature
1. Activation 3 min. 95°C 2. Denaturation 15 sec. 95°C 3. Annealing 10 sec. 60°C 4. Elongation 15 sec. 72°C 35 cycles step 2-4 5. Final 1 min. 72°C
19 3.6.2. Sample preparation
First, the concentrations of gene-patient-specific pools were measured and combination pools were made by combining gene-patient-specific pools with similar molarities. After a clean-up step with magnetic beads, the combination pools were diluted to 1 ng/µL and the library preparation was initiated. Following the Nextera XT (Illumina) protocol, the amplicons of the gene-patient-specific pools were fragmented by transposomes and ligated to adapters in a process called tagmentation. Next, tagmented fragments were PCR amplified with sequencing primer sequences and 2 indices. After another clean-up step, the library DNA was normalized by qPCR, pooled and loaded to the MiSeq sequencer.
3.6.3. Sequencing
The MiSeq sequencer utilizes the Illumina sequencing by synthesis technology. The library is first loaded into a flow cell. After hybridization of the fragments to the flow cell surface, fragments are amplified into a clonal cluster trough bridge amplification. This process is called cluster generation. During each sequencing cycle one of four fluorescently labeled deoxy-ribonucleotide triphosphates (dNTPs) are built into growing DNA strands. Upon incorporation, the specific nucleotide can be identified by its fluorescent signal. The label also functions as a polymerisation terminator, which prevents further dNTPs to be incorporated. This means that only one dNTP signal is sent at a time. The terminator is removed again in the next cycle and the process is repeated. With this method, errors are reduced by natural competition, as in each cycle all four reversible terminator-bound dNTPs are present. NGS enables sequencing on a large scale, as fragments undergo the aforementioned process in a massively parallel way (50).
3.7. Sanger sequencing
With NGS, it is possible that artifacts are wrongly considered as variants (51). To ascertain the authenticity of variants found by the MiSeq Sequencer and thus minimize false-positive results, Sanger sequencing was used. The likelihood of pathogenicity was evaluated for all variants. Only variants expected to be potentially deleterious were selected for Sanger sequencing. 3.7.1. PCR clean-up
Prior to sequencing, the PCR products were cleaned in the post-PCR laboratory. For this step an enzyme solution was needed. The components of the enzyme solution and the volumes used for 20 PCR reactions are listed in table 5. From each PCR product 5 µl was taken and mixed with 1 µl of the enzyme solution. This mixture was then put in a thermocycler, in which an incubation (15 min. 37°C) and inactivation (20 min. 80°C) step were completed.
20 3.7.2. Sequencing
The BigDye® Terminator Cycle Sequencing kit (Applied Biosystems), which is based on the Sanger method, was used. This method requires a denaturated DNA strand, a primer, DNA polymerase, dNTPs and fluorochrome labelled dideoxy-NTPs (ddNTPs), which are called BigDye® Terminators. DNA polymerase will extend the primer attached to a DNA strand, by incorporating dNTPs which are complementary to the ones in the DNA strand. When a dNTP is incorporated, the newly formed complementary strand can be further extended. Alternatively, when a ddNTP is incorporated, further strand extension is blocked, because ddNTPs lack the 3'-hydroxyl group, preventing formation of a phosphodiester bond with the next nucleotide. This results in the formation of fragments of varying length, all ending with a ddNTP (52). In order to sequence in both directions, two separate master mixes were made, one containing a universal M13-forward primer (forward master mix) and the other a universal M13-reverse primer (reverse master mix). The other components were identical for both master mixes and are listed in table 6.
Table 6. Master mix components for one cycle sequencing reaction
Component Volume per reaction
Ready reaction mix (Applied biosystems) 0.5 µl
ABI-buffer (Applied biosystems) 2 µl
Forward/Reverse primer (1µM) 2 µl
Dimethyl sulfoxide 0.5 µl
Water 4 µl
For each reaction, 9 µL of master mix was added to 2 µL of PCR product. The cycling conditions of the reactions are listed in table 7.
Table 7. Conditions for cycle sequencing reactions
Step Time Temperature
1. Activation 5 min. 95°C 2. Denaturation 10 sec. 95°C 3. Annealing 5 sec. 55°C 4. Elongation 4 min. 60°C 25 cycles step 2-4 5. Final 10 min. 15°C
Table 5. Components of enzyme solution used for 20 PCR products
Component Volume
Exonuclease I 1µl
Antartic Phosphatase 4 µl
21 3.7.3. Sanger clean-up
After the sequencing reaction, the DNA fragments were purified using magnetic beads, to remove unincorporated BigDye® Terminators that may interfere with the downstream sequence detection process. First, 10 µL of clean DTR beads (GC Biotech) was added followed by 45 µL of 85% ethanol to each reaction and mixed by pipetting up and down. Next, the reactions were placed on a magnet for 3 minutes, after which supernatant was removed without taking away the beads. The beads were washed twice using 100 µl ethanol (85%). After removal of supernatant, the beads were dried for 10 min at room temperature. The beads were removed from the magnet and resuspended in 40 µl of demineralized water, to eluate the DNA-fragments out of the beads. Finally, the product was placed on the magnet one last time and 30 µl of beadless product was then transferred for sequencing analysis.
3.7.4. Evaluation of Sanger sequencing results
An initial quality check, using A Plasmid Editor (ApE), was performed to see if the Sanger sequencing results were suitable for interpretation. Sequence Patient (SeqPatient), a module of the Sequence Pilot (SeqPilot) software, was used for the actual analysis of the Sanger sequencing data. Within this module result sequences were shown as electropherogram curves, consisting of colored peaks corresponding to the four different bases. For each amplicon two sequences were displayed, a forward one and a reverse one. The reverse sequence functions as a double check. Above the result sequences, a reference sequence was shown. The location within the exon of each base was displayed underneath the result sequences. This made it possible to track the detected variants, which were listed, and to compare them to the reference sequence. It was determined for each variant whether it was an actual variant or an artifact registered by NGS. An artifact can occur when the bases which precede and succeed the suspected variant interfere with the peak which represents the base of interest, leading to the impression of a different base at this position (53).
3.8. Evaluation of variants
To determine whether a variant was potentially pathogenic and thus eligible for confirmation with Sanger sequencing, a number of different factors were considered. Variants that had already been reported, had a reference single nucleotide polymorphism ID number (rs number) and could be looked up on the Single Nucleotide Polymorphism Database (dbSNP). On this database the consequence of each variant was consulted. Possible consequences include: synonymous, missense, indel, intron and splice region. An interpretation of the clinical significance of a variant, accompanied with a review status (rated out of 4), was displayed in ClinVar. Variant interpretations range from benign to pathogenic and are based on assertions of clinical significance submitted by clinical testing laboratories, research laboratories, locus-specific databases, expert panels, etc. Both databases are hosted by the National Center for Biotechnology Information (54).
22 Furthermore, the AF, which was consulted on Genome Aggregation Database (GnomAD), was considered. This was regarded as the most important factor when deciding whether or not to perform Sanger sequencing. Despite the fact that patients participating in this study registered at a European health center, allele frequencies as observed in the total population rather than the frequencies in the European population were used. This can be justified as the origin of the patients who were included in this study was not disclosed. The threshold for possible pathogenicity was arbitrary set at a frequency of 0.10. The chosen threshold is relatively low as sICAD and sVAD are probably non-monogenic diseases, and identified variants could act as influencing genetic risk factors. Variants which were not included in the three above mentioned databases, and therefore have not yet been reported, are considered novel variants. In this study, such novel variants were considered as more likely of being pathogenic. For each variant the number of patients, in which given variant was found, was registered. This data was used to calculate the allele frequency as observed in the study population. Allele frequencies found in this study were compared to allele frequencies as reported in GnomAD when available. However, no statistical analysis was performed as the size of this study sample does not allow meaningful statements regarding the difference in both frequencies.
Locations of identified variants within the protein structure of Notch1 were determined on the basis of the Universal Protein Resource (UniProt), which contains information about protein sequences. This allowed to link variants with the corresponding protein domain of Notch1. Although no information was available about the exact locations of the HD, RAM, PEST and TAD domain. Literature was used to find out if variants that were located in the vicinity of these domains were actually part of them (55–58).
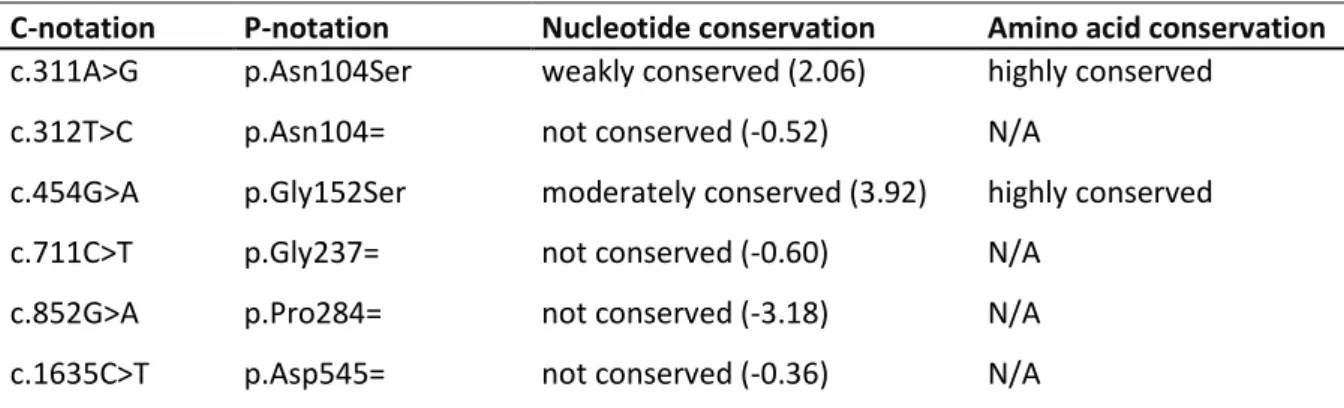
Further evaluation was done through Alamut Visual software version 2.11 (Interactive Biosoftware), which gives access to several prediction tools. Predictions by PhyloP were used to evaluate the evolutionary conservation of nucleotides. PhyloP compares the substitution rate of nucleotides at a certain site to the rate expected under neutral drift. Scores given by PhyloP range between -14.1 and 6.4. Increases in substitution rate at a site, indicating acceleration, are assigned a negative score. While reductions in rate, indicating conservation, are assigned a positive score (59). Alamut Visual uses alignment of orthologues, as provided by Ensembl. This way, amino acids at a certain position are compared across 12 species, which allows interpretation concerning the conservation of given amino acid.
23 Missense variant pathogenicity prediction tools consulted in this study include SIFT, Polyphen-2, Mutation Taster and Align-GVGD. In silico predictions are generated via a scoring system specific to each tool. SIFT scores range from 0.0 (deleterious) to 1.0 (tolerated), with 0.05 as a cut-off value (60). Polyphen-2 classifies variants into “benign” (0.0-0.15) and “possibly damaging” (0.15-1.0) (61). Mutation Taster predicts variants as either “disease causing” or “polymorphism”, the latter means the variant is probably not deleterious. Finally, Align-GVGD scores range from “C0” (least likely deleterious) to “C65” (most likely deleterious). These scores are calculated by combining the Grantham Variation (GV), which is based on differences in biochemical characteristics of amino acids and the Grantham Deviation (GD), which is based on the conservation of an amino acid across species (60). Depending on the outcomes of these 4 prediction tools, the terms “not deleterious”, “conflicting” and “deleterious” were assigned to the variants. When 3 or more prediction tools indicated that a variant is likely to be disruptive, the variant was assigned the term “deleterious”, conversely when 3 or more prediction tools indicated that a variant is probably harmless, it was assigned the term “not deleterious”. Variants were assigned the term “conflicting” when no majority was reached. An additional classification method, based on guidelines by the American College of Medical Genetics and Genomics (ACMG), was applied for all missense variants. This way the following classes were assigned to missense variants: “benign” (class 1), “likely benign” (class 2), “uncertain significance” (class 3), “likely pathogenic” (class 4) and “pathogenic” (class 5). The term likely indicates a 90% certainty (62).
Possible impact of splicing was checked for variants to which it applies. Alamut Visual displayed splicing predictions for some variants. Possible affection on splice sites was then further evaluated for these variants with Human Splicing Finder (HSF).
This study used the Genome Reference Consortium human genome build 37 (GRCh37), also known as hg19, as a genomic reference sequence. As to the coding DNA reference sequence, RefSeq gene transcript sequence NM_017617.3 for NOTCH1 was used, while for the Notch1 protein, the RefSeq protein sequence NP_060087.3 was used.
24
4. Results
4.1. Patient characteristics
In this study, DNA samples of 49 patients with unexplained sICAD and/or sVAD were examined for the presence of NOTCH1 variants. The male-female distribution of these patients was 20 males to 29 females. The mean age at inclusion was 46 years for the male patients and 43 years for the female patients (Table 8). Age, gender and DNA number for each patient are listed in the supplemental section (Supplemental table 3). The following phenotypes of sCAD were present in patients participating in this study: unilateral sICAD (25 patients), bilateral sICAD (3 patients), recurrent sICAD (2 patients), unilateral sVAD (12 patients), bilateral sVAD (4 patients) and co-occurring sICAD and sVAD (3 patients). A table indicating for each patient which variants were identified by NGS in the NOTCH1 gene of given patient is added in the supplemental section (Supplemental table 3). All patients had at least one variation in the sequence of their NOTCH1 gene as investigated with NGS. Only 3 patients had just a single
NOTCH1 variant. The average number of NOTCH1 variants per patient was 7.
Table 8. Demographic characteristics of patients included in this study
Number of patients Mean age at inclusion
Male 20 29 49 46 Female 43 Total 44
4.2. Identified NOTCH1 variants
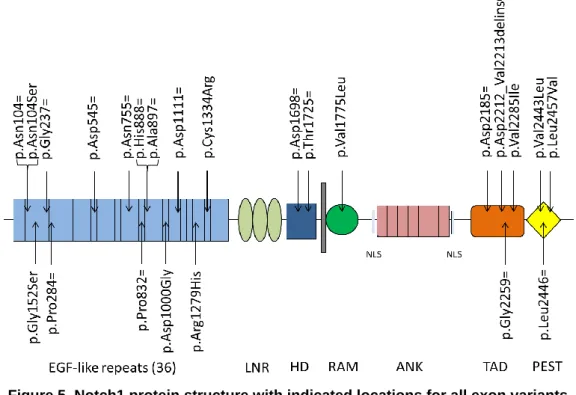
In these patients, 43 different variants were identified by NGS. Sanger sequencing was attempted for 24 variants. This allowed confirmation for only 6 of them, of which 1 was confirmed in 2 patients. The remaining variants were either not confirmed (7 variants) or the Sanger sequencing results were insufficient to allow interpretation (11 variants). The variant which was confirmed for 2 patients, was present in a homozygous way in 1 of them. All other confirmed variants were present in a heterozygous way (Table 9). Sanger reports of confirmed variants are added in the supplemental section (Supplemental figure 2). The majority of these variants has already been reported and was thus present in dbSNP, GnomAD and ClinVar. Rs numbers were not available for 10 variants, meaning these are novel variants which are not yet reported (Table 9). Alamut Visual reports of each variant are added in the supplemental section (Supplemental figure 3).
25
C-notation P-notation G-notation Exon Intron Rs number AF
(GnomAD) AF (Study)
# patients
Type ClinVar Zygosity Sanger
c.311A>G p.Asn104Ser g.139418261T>C 3 rs199654211 0.0011 0.041 2 missense likely benign ** HE/HO C
c.312T>C p.Asn104= g.139418260A>G 3 rs4489420 0.59 0.65 32 synonymous benign ** /
c.454G>A p.Gly152Ser g.139417590C>T 4 rs750242131 0.000050 0.020 1 missense uncertain significance * /
c.711C>T p.Gly237= g.139417333G>A 4 rs61751557 0.0075 0.020 1 synonymous benign ** /
c.852G>A p.Pro284= g.139413908C>T 5 rs2229975 0.13 0.27 13 synonymous benign * /
c.1635C>T p.Asp545= g.139410467G>A 10 rs11574889 0.0079 0.041 2 synonymous benign ** F
c.2265T>C p.Asn755= g.139407932A>G 14 rs2229971 0.41 0.45 22 synonymous benign * /
c.2496G>T p.Pro832= g.139405695C>A 16 rs61751551 0.00059 0.020 1 synonymous benign/likely benign ** /
c.2664C>T p.His888= g.139405181G>A 17 rs61751548 0.012 0.020 1 synonymous benign ** F
c.2691C>T p.Ala897= g.139405154G>A 17 rs11574895 0.012 0.020 1 synonymous benign ** F
c.2999A>G p.Asp1000Gly g.139403494T>C 19 N/A N/A 0.020 1 missense N/A F
c.3333C>T p.Asp1111= g.139402584G>A 21 rs61751545 0.0039 0.020 1 synonymous benign/likely benign ** HE C
c.3836G>A p.Arg1279His g.139401233C>T 23 rs61751543 0.016 0.020 1 missense benign ** NC
c.4000T>C p.Cys1334Arg g.139400993A>G 24 N/A N/A 0.020 1 missense N/A HE C
c.5094C>T p.Asp1698= g.139397707G>A 27 rs10521 0.45 0.63 31 synonymous benign * /
c.5175C>T p.Thr1725= g.139396933G>A 28 rs61751536 0.0026 0.020 1 synonymous benign/likely benign ** HE C
c.5323G>C p.Val1775Leu g.139396785C>G 28 N/A N/A 0.020 1 missense N/A NC
c.6555C>T p.Asp2185= g.139391636G>A 34 rs2229974 0.62 0.86 42 synonymous benign ** /
c.6636_6637delCGinsAC p.Asp2212_Val2213delinsGluLeu g..139391554_139391555delinsGT 34 N/A N/A 0.020 1 indel N/A NC
c.6777T>C p.Gly2259= g.139391414A>G 34 rs61751490 0.0019 0.020 1 synonymous benign ** /
c.6853G>A p.Val2285Ile g.139391338C>T 34 rs61751489 0.029 0.041 2 missense benign ** F
c.7327G>T p.Val2443Leu g.139390864C>A 34 N/A N/A 0.020 1 missense N/A F
c.7338G>A p.Leu2446= g.139390853C>T 34 rs35320927 0.00090 0,020 1 synonymous benign/likely benign ** /
c.7369C>G p.Leu2457Val g.139390822G>C 34 rs61755043 0.00058 0.020 1 missense benign/likely benign ** F
c.61+9C>T N/A g.139440169G>A 1 N/A N/A 0.020 1 intron N/A NC
c.61+18C>T N/A g.139440160G>A 1 N/A N/A 0.020 1 intron N/A /
c.1100-8C>T N/A g.139412752G>A 6 rs545088400 0.00036 0.020 1 splice region conflicting* HE C
26
C-notation P-notation G-notation Exon Intron Rs number AF
(GnomAD) AF (Study)
# patients
Type ClinVar Zygosity Sanger
c.1100-16G>C N/A g.139412760C>G 6 rs146815871 0.0038 0.020 1 intron benign * /
c.1441+7C>T N/A g.139412197G>A 8 rs9411208 0.58 0.73 36 splice region benign /
c.1555+10A>G N/A g.139411714T>C 9 rs11145767 0.46 0.55 27 intron benign ** /
c.1555+15C>T N/A g.139411709G>A 9 rs577175237 0.000056 0.020 1 intron N/A NC
c.1555+15delinsTTGT N/A g.139411709delinsACAA 9 N/A N/A 0.020 1 intron N/A F
c.1555+16G>A N/A g.139411708C>T 9 rs368798926 0.000040 0.041 2 intron likely benign * F
c.1555+17C>T N/A g.139411707G>A 9 N/A N/A 0.020 1 intron N/A F
c.1669+9T>C N/A g.139410424A>G 10 rs3125006 0.87 0.96 47 intron benign * /
c.1670-9A>G N/A g.139410177T>C 10 rs3124603 0.47 0.55 27 intron benign ** /
c.2353+16G>A N/A g.139407828C>T 14 rs771973233 0.000032 0.020 1 intron N/A F
c.2587+20G>A N/A g.139405584C>T 16 rs148381982 0.017 0.020 1 intron benign* /
c.2588-4G>A N/A g.139405261C>T 16 rs3125001 0.48 0.51 25 splice region benign ** /
c.2740+12C>T N/A g.139405093G>A 17 rs36119806 0.11 0.20 10 intron benign * /
c.5639-12C>T N/A g.139395311G>A 30 rs11574908 0.067 0.12 6 intron benign ** HE C
c.6181-16delC N/A g.139392026del 33 N/A N/A 0.020 1 intron N/A NC
c.6181-18C>T N/A g.139392028G>A 33 rs1257719720 0.0000055 0.020 1 intron N/A NC
C-notation: Coding DNA sequence (RefSeq: NM_017617.3), P-notation: Protein sequence (RefSeq: NP_060087.3), G-notation: Genomic DNA sequence (Build: GRCh37), Rs-number: Reference single nucleotide polymorphism ID number, AF (Gnomad): Allele frequency as reported in GnomAD, AF (study): Allele frequency as observed in the study population, # patients: Number of patients in which the variant was identified, Sanger: indicates whether or not a variant was confirmed by Sanger sequencing, N/A: Not available, C: The variant was confirmed by Sanger sequencing, NC: The variant was not confirmed by Sanger sequencing, F: Sanger sequencing was attempted, but failed, /: No sanger sequencing was attempted.