FOLLOW-UP VAN KINDEREN
MET SIKKELCELZIEKTE
Delphine Gryson
Stamnummer: 00805500Promotor 1: Dr. Julie Willekens Promotor 2: Dr. Bram De Wilde
Masterproef master in de specialistische geneeskunde
III
Voorwoord
Deze masterproef werd tot stand gebracht in het kader van het opleidingsonderdeel Masterproef binnen de opleiding Master in de specialistische geneeskunde, afstudeerrichting Pediatrie aan de faculteit Geneeskunde en gezondheidswetenschappen van de Universiteit Gent.
Zonder steun en hulp van een aantal mensen zou deze masterproef niet tot stand gekomen zijn. Ik wil dan ook graag een bijzonder woord van dank richten aan mijn promotoren, Dr. Julie Willekens en Dr. Bram De Wilde, voor hun begeleiding en waardevolle advies bij deze masterproef.
Tenslotte een speciaal woordje van dank aan mijn familieleden, vrienden en medestudenten. Hun ondersteuning, moreel en praktisch, is van onschatbare waarde geweest bij het schrijven van deze masterproef.
IV
Inhoud
Abstract ... 1 Inleiding ... 2 Methodologie ... 3 Resultaten ... 4 1 Epidemiologie ... 4 2 Pathofysiologie ... 4 3 Diagnostiek ... 5 3.1 Neonatale hielprikscreening ... 5 3.2 Familieonderzoek ... 6 3.3 Klinische symptomen ... 63.4 Afwijkende bevindingen bij bloedonderzoek ... 6
4 Complicaties ... 6 4.1 Pijn ... 6 4.2 Infecties en koorts ... 7 4.3 Cerebrovasculaire aandoeningen ... 8 4.4 Anemie ... 8 4.5 Oftalmologische aandoeningen ... 9 4.6 Nefrologische aandoeningen ... 10 4.7 Cardiopulmonaire aandoeningen ... 10 4.8 Hepatobiliaire aandoeningen... 12
4.9 Genito-urinaire aandoeningen, ontwikkeling en seksualiteit ... 13
4.10 Osteoarticulaire aandoeningen ... 14
4.11 Ulcus cruris ... 14
4.12 Psychosociaal ... 14
4.13 Conclusie ... 15
V
5.1 Eerste bezoek en opvolgbezoeken ... 15
5.2 Screening en preventie van complicaties ... 17
5.3 Behandeling ... 27
5.4 Perioperatieve zorg ... 30
5.5 Overgang naar volwassenheid ... 30
Discussie ... 30 Referentielijst ... 31 Appendix ... 42 1 Complicaties ... 42 2 Indicatie hydroxyureumtherapie... 43 3 Eerste bezoek ... 44 4 Routine-evaluatie ... 44 5 Leeftijdsgebonden follow-up ... 46 6 Figuren ... 48
VI
Afkortingen
ACS Acute chest syndrome
AV Avasculaire necrose
CDC Centers for Disease Control and Prevention cGMP Cyclisch guanosinemonofosfaat
CRVO Centrale Retinale veneuze occlusie
CVA Cerebrovasculair accident
HbAS Hemoglobine A + Hemoglobine S
HbS Hemoglobine S
HbSC Hemoglobine S + Hemoglobine C
HbSS Homozygotie voor HbS
HLA Humaan leukocytenantigeen
mPAP Gemiddelde arteriële druk ter hoogte van de arteria pulmonalis MRA
MRI
Magnetic Resonance Angiography Magnetic Resonance Imaging
NHLBI National Heart, Lung, and Blood Institute OSAS Obstructief slaapapneusyndroom
PH Pulmonale hypertensie
PSR Proliferatieve sikkelretinopathie RHC Katheterisatie van de rechter harthelft
SCA Sikkelcelanemie (HbSS en HbS-β0-thalassemie) SCD Sikkelcelziekte (HbSC en HbS-β+-thalassemie)
SDB Sleep-disordered breathing, ademhalingsgebonden slaapstoornissen TCD Transcraniële doppler ultrasonografie
TCV Transcraniële velocity
TRV Tricuspid regurgitation velocity
VOC Vaso-occlusieve crisis
1
Abstract
Achtergrond: Sikkelcelziekte – vroeger een exotische aandoening in West-Europa – is door toenemende migratie geëvolueerd naar een wereldwijd gezondheidsprobleem. Het aantal patiënten met sikkelcelziekte zal blijven toenemen. In welstellende landen is de levensverwachting sterk gestegen door verschillende interventies, waaronder vaccinatie en penicilline profylaxe. Sikkelcelziekte is zo een chronische aandoening geworden, met noodzaak tot goede follow-up en langetermijnbehandeling.
Methode: Er werd eerst een literatuurstudie uitgevoerd om de verschillende complicaties van sikkelcelziekte bij kinderen te identificeren. Op basis van de in kaart gebrachte complicaties werd een nieuw literatuuronderzoek uitgevoerd, dat per complicatie de screening-, preventie-, management- en follow-upmaatregelen onderzoekt. In de literatuurstudies werden Engelstalige reviewartikels, klinische studies en expert panel reports opgenomen.
Resultaat: Sikkelcelziekte omvat vier belangrijke genotypes, met een verschillend ziektebeeld. HbSS en HbS-β0-thalassemie, samen ‘sikkelcelanemie’, kennen het meest ernstige verloop.
HbSC en HbS-β+-thalassemie zijn milder. Patiënten met sikkelcelziekte ervaren een breed spectrum aan complicaties, zowel acuut als chronisch: pijn, infecties en koorts, cerebrovasculaire aandoeningen, anemie, oftalmologische aandoeningen, nefrologische aandoeningen, genito-urinaire aandoeningen, osteoarticulaire aandoeningen, ulcus cruris en psychosociale moeilijkheden. ACS, stroke, de vaso-occlusieve crisis en aplastische crisis zijn de meest voorkomende complicaties. Bij de follow-up van kinderen met sikkelcelziekte is aandacht voor de verschillende screeningmaatregelen die deze in een vroeg stadium kunnen identificeren (bloed- en urine-analyse, transcraniële doppler, elektrocardiografie, ….) alsook voor de educatie van de patiënt en zijn familie in het management en de preventie hiervan (profylactisch gebruik van penicilline, vaccinaties, vermijden triggers, ..) heel belangrijk. Daarnaast kunnen ook een aantal ziekte-modificerende therapieën de ziekte milderen (hydroxyureumtherapie, transfusietherapie) of genezen (stamceltransplantatie en in de toekomst gentherapie). Dankzij deze aanpak (preventie, management en behandeling) is de levensverwachting van kinderen met sikkelcelziekte sterk toegenomen.
Conclusie: Op basis van de beschikbare literatuur werd een voorstel geformuleerd voor een vernieuwd protocol voor de opvolging van kinderen met sikkelcelziekte, zowel bij het eerste bezoek als bij de routine-evaluaties. Deze zijn enerzijds evidence-based, en berusten anderzijds op de heersende consensus.
2
Inleiding
Sikkelcelziekte is wereldwijd een toenemend gezondheidsprobleem (1). Door toenemende migratie is sikkelcelziekte geen exotische aandoening meer in West-Europa (2).
Het aantal patiënten met sikkelcelziekte zal verder toenemen, zowel in welstellende landen als in de onderontwikkelde landen. In welstellende landen is de levensverwachting sterk gestegen door verschillende interventies zoals neonatale screening, vaccinatie, penicilline profylaxe, primaire beroerte preventie en behandeling met hydroxyureum (3).
De mediane overleving bedraagt nu meer dan 60 jaar en kindersterfte is vergelijkbaar met deze van de algemene populatie (1, 3). In ontwikkelde landen bereiken meer dan 95% van de kinderen met sikkelcelziekte volwassenheid (3). In Afrika, waar er een gebrek is aan neonatale screening, vaccinatie en waar malaria, ondervoeding en armoede grote uitdagingen zijn, bedraagt de mortaliteit bij kinderen met sikkelcelziekte jonger dan 5 jaar tot 90%.
Recent werden enkele grootschalige screeningsprogramma’s gelanceerd met als gevolg dat ook in onderontwikkelde landen een toenemend aantal baby’s en jonge kinderen overleven tot volwassen leeftijd. Echter maakt het gebrek aan basis gezondheidszorginfrastructuur in vele regio’s de preventie en behandeling van sikkelcelziekte aldaar heel moeilijk (1).
Sikkelcelziekte is een chronische, complexe multisysteemaandoening die een aanpak vraagt met daarin een rol voor screening, preventie, educatie en management van zowel acute als chronische complicaties binnen het kader van een allesomvattend zorgprogramma (4). Een goede follow-up van kinderen met sikkelcelziekte, van bij de diagnose, is zeer belangrijk. Het doel van deze literatuurstudie is het samenbrengen van de beschikbare kennis hieromtrent in een protocol voor de follow-up van sikkelcelziekte bij kinderen.
3
Methodologie
De uitgebreide literatuurstudie werd uitgevoerd op de online databases PubMed, Web of Science, Cochrane Library en Google Scholar.
In een eerste fase, bij het identificeren van de verschillende complicaties van sikkelcelziekte bij kinderen, werden volgende zoektermen gebruikt: “pediatric sickle cell”, “sickle cell disease children”, “sickle cell anemia children”, “sickle cell complications”, “sickle cell complications children”, “follow-up sickle cell children”, “pediatric follow-up sickle”, “screening sickle cell children”, “pediatric screening sickle cell”, “management sickle cell children”, “pediatric sickle cell”, “complications sickle pediatric, “acute sickle pediatric”, “chronic sickle pediatric”, “comprehensive care sickle children” en andere gelijkaardige woordcombinaties.
Na de analyse van de literatuur over de verschillende acute en chronische complicaties van sikkelcelziekte bij kinderen, alsook over de algemene follow-up en het management van sikkelcelziekte, werd gezocht naar literatuur over screening, preventie, management en follow-up van één bepaalde complicatie, behandeling, screeningmethode of -parameter, bijvoorbeeld:
• “sickle cell children acute chest syndrome” • “sickle cell pediatric acute chest syndrome” • “sickle cell transcranial doppler children”
• “pediatric sickle cell tricuspid regurgitant velocity” • “sickle cell children bilirubin”
• “sickle cell pediatric sleep apnoea” • “sickle pediatric perioperative” • “hydroxycarbamide sickle children” • “hydroxyurea sickle children” • “transfusion sickle pediatric” • …
Literatuur werd geëvalueerd op haar kwaliteit met behulp van de impactfactoren. Zowel reviewartikels, expert panel reports als clinical trials werden weerhouden. Individuele cases werden niet weerhouden. Enkel Engelstalige artikels werden weerhouden.
Alle literatuur werd verzameld in een centrale database met behulp van het programma Endnote (versie X9), dat eveneens gebruikt werd voor referenties en de referentielijst in dit document.
4
Resultaten
1 Epidemiologie
Dragerschap voor sikkelcelziekte, sickle cell trait (i.e. heterozygositeit voor de sikkelcelmutatie in HBB) verhoogt de kans om malaria tropica te overleven (1, 2). Door dit selectieve voordeel hebben mutaties in het gen dat codeert voor de β-keten in hemoglobine, resulterend in het sikkelcelallel, zich verspreid in gebieden waar malaria endemisch is: equatoriaal Afrika, het oostelijk deel van het Middellandse Zeegebied, het Arabisch schiereiland en delen van India. Door de enorme migratie in de afgelopen eeuwen, onder andere door de gedwongen ontheemding van miljoenen mensen vanuit West-Afrika naar Amerika gedurende de slavernij en de recente wereldwijde migratiestromen, is sikkelcelziekte niet meer beperkt tot de oorspronkelijke regio’s en wordt ze beschouwd als een aandoening met globale impact (1, 2). Jaarlijks worden wereldwijd ongeveer 300.000 kinderen geboren met sikkelcelziekte (1, 2, 5), de meerderheid in Nigeria, Congo en India (1), met een globaal geschatte incidentie van 112 per 100.000 geboortes (5). In de Verenigde Staten, met een grote Afro-Amerikaanse vertegenwoordiging, wordt het totaal patiënten met sikkelcelziekte geschat op 100.000, met een incidentie van 1550 per 100.000 geboortes (6, 7). In België is de geschatte incidentie minstens 50 op 100.000 geboortes (8), in lijn met het geschatte Europese gemiddelde van 43.12 per 100.000 geboortes (95% CI = 30.31 – 55.92)(5). In september 2018 waren 538 patiënten ingeschreven in het Belgische register voor sikkelcelziekte, waarvan 285 werden geboren in België. Onder hen werden 182 patiënten geïdentificeerd door middel van neonatale hielprikscreening, 53 van hen werden geboren in een Belgisch ziekenhuis zonder screening.
2 Pathofysiologie
Normaal hemoglobine A bestaat uit vier polypeptideketens, twee α-ketens en twee β-ketens. Sikkelcelziekte is het resultaat van een puntmutatie van het β-globinegen op chromosoom 11, wat resulteert in een variant van hemoglobine: sikkelglobine, of hemoglobine S (HbS) (9). Hierdoor wordt het aminozuur glutaminezuur in de β-globineketen vervangen door valine [Glu6Val, rs 334] (1, 2), wat leidt tot de productie van HbS.
Sikkelcelziekte erft autosomaal recessief over en omvat vier belangrijke genotypes, waarvan homozygotie voor HbS (HbSS) met ongeveer 70% de belangrijkste is. Compound heterozygositeit met andere hemoglobinevarianten (HbC, HbF en HbD) zorgt voor andere genotypes en fenotypes van sikkelcelziekte, en het resulterende HbSC (HbS + HbC), is het tweede belangrijke genotype. Er kan ook compound heterozygositeit ontstaan door een
5 combinatie van het HbS-gen en een β-thalassemieallel. Dat geeft aanleiding tot de genotypes HbS-β0-thalassemie en HbS-β+-thalassemie. Bij heterozygotie voor HbS en een normaal
hemoglobine A (HbAS) zijn patiënten drager voor sikkelcelziekte (sickle cell trait), maar vertonen zij geen klinische ziekte. Het dragerschap is wel van belang vanuit klinisch-genetisch oogpunt (1, 2, 9).
De meest ernstige vormen van sikkelcelziekte zijn de genotypes HbSS en HbS-β0-thalassemie, gevolgd door de mildere varianten HbSC en HbS-β+-thalassemie (1, 2, 9). De term sikkelcelziekte wordt gebruikt om alle genotypes te beschrijven die het klassieke klinische syndroom veroorzaken (4). De term sikkelcelanemie wordt gebruikt om naar HbSS en HbS-β0-thalassemie te verwijzen, aangezien ze klinisch een zeer gelijkaardig verloop kennen (10).
Sikkelcelziekte is een chronische, complexe, multi-systeem en -orgaanziekte. Wanneer HbS in voldoende hoge concentratie aanwezig is in de erytrocyt, heeft het de neiging om in een zuurstofarme omgeving te polymeriseren (stress, hypoxie, acidose). De erytrocyten worden stug, fragiel en sikkelvormig en hun gemiddelde levensduur daalt naar slechts 15 tot 20 dagen, in vergelijking met 120 dagen bij een normale erytrocyt. Daarnaast ontstaat interactie tussen sikkelcel en de vaatwand, wat resulteert in vaso-occlusie, ischemie, inflammatie, stollingsactivatie, reperfusieschade en de vorming van nieuwe fragiele bloedplaatjes. De infarctering, vaatschade en bloedingen die hier het gevolg van zijn, resulteren in weefsel- en orgaanschade, wat dan weer aanleiding geeft tot ernstige pijn en orgaanfalen (1, 2, 9).
3 Diagnostiek
3.1 Neonatale hielprikscreening
In België wordt sikkelcelziekte (of sickle cell trait) nog niet opgespoord door middel van een neonataal hielprikscreeningprogramma op nationaal niveau, in tegenstelling tot Nederland waar dit gebeurt sinds 2007. Regionaal zijn wel twee screeningprogramma’s geïntegreerd in Brussel (alle neonaten, alle kraamafdelingen) en Luik (15 materniteiten), samen goed voor 33% van de geboortes in België (8). De kinderen die hiermee worden opgespoord zijn nog asymptomatisch, aangezien er bij de geboorte nog overwegend HbF aanwezig is. Deze wordt in de loop van de eerste 4-6 levensmaanden vervangen door HbA1 (in de normale situatie) of HbS (in het geval van homozygote sikkelcelziekte). Dankzij screening kan met antibiotica profylaxe de morbiditeit en mortaliteit van infecties met gekapselde bacteriën voorkomen worden. Ook oudervoorlichting omtrent belangrijke klinische symptomen kan vroeger worden gestart.
6 3.2 Familieonderzoek
Bij patiënten met een positieve familiale anamnese voor sikkelcelziekte of sickle cell trait, is er een reden om diagnostiek in te zetten. Ook de etnische herkomst van een persoon, zoals mensen met voorouders afkomstig uit gebieden met een hoge dragerschapsfrequentie van hemoglobinopathie (Middellandse Zeegebied, Afrika, Azië, Midden-Oosten, Caribisch gebied) kan een reden zijn om diagnostiek te starten. Bij vermoeden van hemoglobinopathie wordt Hb-elektroforese, eventueel in combinatie met DNA-onderzoek, aangevraagd.
3.3 Klinische symptomen
Volgende symptomen en/of ziektebeelden vragen om diagnostiek naar sikkelcelziekte: langdurige of acuut verergerende bleekheid, icterus of persisterende donkere urine, acute pijn in de ledematen, rug of buik, pneumokokken meningitis, -sepsis en/of -pneumonie, acute splenomegalie, stroke of acute dyspneu met pijn en koorts.
3.4 Afwijkende bevindingen bij bloedonderzoek
Vaak worden bij niet-gerelateerde bloedonderzoeken toevallig aanwijzingen gevonden voor de aanwezigheid van een hemoglobinopathie, zoals afwijkingen passend bij een hemolytische anemie (normocytaire anemie, verhoging LDH, verlaagd haptoglobine, verhoogd bilirubine) of de aanwezigheid van HbS. Ook hiervoor kan diagnostiek naar sikkelcelziekte gestart worden.
4 Complicaties
Patiënten met sikkelcelziekte ervaren een breed spectrum aan complicaties, zowel acuut als chronisch. Dit leidt tot een verminderde levenskwaliteit en een kortere levensverwachting. De complicaties worden ernstiger naarmate de patiënt ouder wordt (9). Veel van deze complicaties volgen uit het sikkelen van de erytrocyten, wat zorgt voor een versnelde afbraak ervan, alsook occlusies in de kleine bloedvaten die leiden tot weefsel- of orgaanschade.
4.1 Pijn
4.1.1 Acute pijn, de vaso-occlusieve crisis
Acute pijn, ook wel ‘vaso-occlusieve crisis’ (VOC) of ‘sikkelcelcrisis’, is de meest voorkomende complicatie bij patiënten met sikkelcelziekte. Een VOC gaat gepaard met plotse pijn zonder enige voorafgaande waarschuwing, meest frequent gelokaliseerd ter hoogte van de beenderen, thoracaal of abdominaal. Bij een VOC treedt infarctering van weefsels op als gevolg van plaatselijke vaatafsluiting door sikkelcelvorming (11). VOC is een klinische diagnose, zonder objectieve diagnostische tests. De pijn wordt omschreven als scherp, intens, stekend of kloppend (11). Bijna alle patiënten met sikkelcelziekte worden hier ooit mee geconfronteerd, al vanaf een leeftijd van 3 - 4 maanden, wanneer het foetale hemoglobine vervangen is door
7 HbS. Dactylitis, een pijnlijke ontsteking van hand- en/of voetbotjes door infarctering, is vaak de eerste manifestatie van sikkelcelziekte bij jonge kinderen. De crisis kan getriggerd worden door verblijf op hoogte, dehydratatie, ziekte, stress of temperatuursveranderingen (9, 11). 4.1.2 Chronische pijn
Chronische pijn, pijn die langer dan 3 maanden aanhoudt (12), is eveneens een vaak voorkomende complicatie van sikkelcelziekte, voornamelijk bij adolescenten en volwassenen. Deze pijn is moeilijker te omschrijven en beschrijvingen lopen uiteen: diep en dof, of neuropathisch met een brandend gevoel en tintelingen. Verschillende comorbiditeiten van sikkelcelziekte, bv. avasculaire necrose, kunnen mee aan de basis liggen (12). Chronische pijn kan versterkt worden door -en versterkt- depressie, angststoornissen en andere psychosociale ziektebeelden (zie ook 4.12 Psychosociaal). De oorzaak van chronische pijn is een domein van verder onderzoek en wordt momenteel nog onvoldoende begrepen (10).
4.2 Infecties en koorts
Herhaalde, vaak subklinische, miltinfarcten resulteren bij de meerderheid van de kinderen met sikkelcelanemie (HbSS en HbS-β0-thalassemie) al in het eerste levensjaar in een functionele asplenie. Dat leidt tot een verhoogd risico op infecties met gekapselde bacteriën (vnl. pneumokokken, Haemophilus influenza type B en meningokokken) en deze kwetsbaarheid neemt verder toe door weefselnecrose. Infecties dragen wereldwijd bij tot de vroege mortaliteit bij sikkelcelziekte (2). Hoewel de incidentie hiervan gedaald is door het profylactisch gebruik van penicilline en vaccinatie tegen pneumokokken (13), moeten alle gevallen van koorts bij patiënten met sikkelcelziekte beschouwd worden als een urgentie omdat het kan gaan over penicilline-resistente organismen of de vaccinatiestatus van de patiënt onbekend is (10). Bij kinderen met HbSC en HbS-β+-thalassemie is er een veel minder groot risico op levensbedreigende infecties, omdat hun miltfunctie normaal is of minimaal aangetast wordt tijdens hun kinderjaren. Oudere kinderen en volwassenen met alle SCD-genotypes lopen wel een hoger risico op een invasieve bacteriële infectie, zoals invasieve pneumokokkenziekte (10). Koorts kan ook een symptoom zijn van andere levensbedreigende aandoeningen, zoals acuut coronair syndroom, bv. door een infectie met mycoplasma. Patiënten met sikkelcelziekte zijn ook at risk voor gramnegatieve enterische infecties, infecties van de urinewegen, het hepatobiliaire systeem. Koorts kan ook een indicatie zijn van osteomyelitis, veroorzaakt door staphylococcus aureus, salmonella of andere enterische pathogenen (10).
8 4.3 Cerebrovasculaire aandoeningen
Patiënten met sikkelcelziekte hebben een hoger risico op vasculaire complicaties, waar het cerebrovasculair accident (CVA) de belangrijkste van is. Symptomen zijn evenwichtsstoornissen, afhangend gelaat, ernstige hoofdpijn, stuipen, afasie of hemiparese. Bij kinderen komen CVA’s het vaakst voor tussen de leeftijd van 2 en 9 jaar (10). Nog voor de leeftijd van 20 jaar, worden maar liefst 11% van alle patiënten met sikkelcelanemie (HbSS en HbS-β0-thalassemie) getroffen door een ischemische of hemorragische beroerte (14).
Deze patiënten worden ook vaak getroffen door een minder uitgesproken vorm, de silent stroke of stille beroerte (asymptomatische micro-infarctering), waarbij geen symptomen optreden maar waarbij op een MRI wel een cerebrale ischemie detecteerbaar is (9). Deze is te herkennen aan slechte of afnemende schoolprestaties, ontwikkelingsachterstand of leerstoornissen (10). Stille beroertes zijn een belangrijke complicatie, met een risico van 11% (leeftijd van 15 maanden) tot 27.7% (leeftijd van 3 – 4 jaar) bij kinderen met HbSS en 5% tot 13.5% bij kinderen met HbSC (15-17). In nieuw onderzoek van Ogunsile et al. (2018, n=958)(18) worden stille infarcten gelinkt aan een eerdere besmetting met het parvovirus en lage Hgb-waarden.
Een klinische beroerte wordt geassocieerd met een afname van 10 – 15 punten op de IQ-schaal, stille infarcten met een verlies van 5 punten (19).
4.4 Anemie
De meeste patiënten met sikkelcelziekte lijden aan een milde tot matig ernstige chronische vorm van anemie. In sommige gevallen kan dit evolueren naar een levensbedreigende vorm.
Acute anemie wordt gedefinieerd als een vermindering van minstens 2.0 g/dL tegenover de baseline waarde. Deze is anders voor elke patiënt en onder meer afhankelijk van het hemoglobine genotype, voorbije en huidige behandelingen en andere onbekende factoren. Typische baselinewaarden zijn 6 – 8 g/dL voor patiënten met HbSS en HbS-β0-thalassemie, 9
– 12 g/dL voor patiënten met HbS-β+-thalassemie en 10 – 15 g/dL voor patiënten met HbSC.
Acute anemie is vaak het gevolg van een aplastische crisis of miltsequestratie (10). 4.4.1 Aplastische Crisis
Bij een aplastische crisis stopt het beenmerg met het aanmaken van nieuwe erytrocyten. Symptomen zijn vermoeidheid, bleekheid, kortademigheid en soms zelfs syncopes. Bij klinisch onderzoek zal vaak lethargie en een tachycardie worden opgemerkt (10, 12).
De crisis wordt vaak veroorzaakt door een parvovirus B19-infectie, of vijfde kinderziekte. De infectie verspreidt zich meestal via de luchtwegen, maar overdracht kan ook via de placenta of
9 bloed. Patiënten vertonen grieperige symptomen en erythema infectiosum. De ziekte is makkelijk te verwarren met rubella, of andere exantheemziektes (20). De infectie is normaal gezien mild van aard, maar kan voor patiënten met sikkelcelziekte voor ernstige complicaties zorgen, aangezien bij deze infectie de aanmaak van erytrocyten in het beenmerg gedurende zeven tot tien dagen onderdrukt wordt. Aangezien de gemiddelde levensduur van een sikkelcelerytrocyt 15 tot 20 dagen is, kan deze situatie aanleiding geven tot een acute, soms levensbedreigende anemie (2, 3, 10, 12), met typische waarden van < 1% voor de reticulocyten (12).
4.4.2 Miltsequestratie
Miltsequestratie is eveneens een belangrijke oorzaak van acute anemie bij kinderen met sikkelcelanemie. Deze gaat gepaard met pooling van erytrocyten, waardoor de milt sterk opzwelt en de hoeveelheid beschikbare erytrocyten in het lichaam daalt. Hierdoor ontstaat anemie. Miltsequestratie wordt gedefinieerd als een sterke zwelling van de milt en een reductie van minstens 2 g/dL onder de baselinewaarde voor hemoglobineconcentratie (10, 21).
Miltsequestratie ontwikkelt zich zonder enige waarschuwing of oorzaak en kan al plaatsvinden als het kind enkele maanden oud is, hoewel de ziekte meer voorkomt bij kinderen tussen 1 en 4 jaar. Bij kinderen met HbSS heeft vaak involutie en auto-infarctering van de milt al plaatsgevonden voor het vijfde levensjaar, waardoor sequestraties vanaf die leeftijd minder plaatsvinden. Bij zuigelingen met HbSS kunnen deze zich zeer acuut voordoen in combinatie met ernstige anemie en een hypovolemische shock. Bij patiënten met HbSC en HbSβ+ -thalassemie kan miltsequestratie later of op volwassen leeftijd nog plaatsvinden (10).
4.5 Oftalmologische aandoeningen 4.5.1 Chronisch
Ook de bloedvaten in het oog kunnen worden aangetast door sikkelcelziekte, waarbij de retina het vaakst getroffen wordt en retinopathie dus de meest voorkomende oogaandoening is (10). De meest ernstige vorm van retinopathie is proliferatieve (sikkel) retinopathie (PSR), omdat haar progressie gepaard gaat met verminderd gezichtsvermogen. De prevalentie van proliferatieve retinopathie is 45% bij patiënten met HbSC, 11% bij patiënten met HbSS en 17% van de patiënten met sikkel β-thalassemie (22), nog voor volwassenheid wordt bereikt. De piekprevalentie ligt vroeger bij HbSC (15 – 24 jaar bij mannen en 20 – 39 jaar bij vrouwen) (10). De onvermijdelijke retinale ischemie en neovascularisatie die hierdoor ontstaan, kan leiden tot netvliesloslating en verlies van gezichtsvermogen (10, 12).
10 4.5.2 Acuut
Patiënten worden daarnaast ook geconfronteerd met acute oogaandoeningen die vaak volgen op trauma, infectie of een VOC. Deze complicaties leiden tot occlusie van het vaatstelsel rond de ogen of een versnelde progressie van PSR (10). Ze kunnen verregaande gevolgen hebben voor de patiënt, waaronder permanent verlies van gezichtsvermogen. Hyfemie, retinale veneuze occlusie (CRVO), orbitale en periorbitale infecties, orbitale infarcten, netvliesloslating en orbitaal compartimentsyndroom vragen allen dringend medisch ingrijpen om dat te voorkomen. 4.6 Nefrologische aandoeningen
4.6.1 Chronisch
Sikkelcelnefropathie, de aantasting van de nieren door het sikkelen van de erytrocyten, is ook een belangrijke complicatie en veroorzaakt tubulaire en medullaire dysfunctie. Er is weinig onderzoek naar de effecten hiervan op de morbiditeit en mortaliteit bij kinderen, aangezien de meeste dergelijke complicaties zich voordoen bij volwassenen (23). Microscopische hematurie komt veel voor bij pediatrische sikkelcelnefropathie, met een prevalentie tot 30% (23).
De meest voorkomende complicatie bij kinderen is hyposthenurie, waarbij er moeilijkheden zijn om de urine te concentreren, wat leidt tot frequent urineren (10, 12). Hierdoor hebben deze patiënten ook een hoger risico op hypovolemie, omdat het lichaam niet op een verminderde inname van vloeistoffen kan reageren door de urine te concentreren. Ook enuresis is een frequente complicatie ten gevolge van hyposthenurie, met een prevalentie tot 42 procent bij kinderen van 6 - 8 jaar en 9 procent bij volwassenen tussen de leeftijd van 18 – 20 jaar.
Bij het ouder worden evolueren de nefrologische complicaties naar microalbuminurie, proteïnurie, glomerulosclerose en chronisch nierfalen (10, 12). Proteïnurie heeft een prevalentie van 6.2 tot 41% bij kinderen, chronische nierinsufficiëntie 8% tot 26.5% (24, 25).
4.6.2 Acuut
Acuut nierfalen vindt vaak plaats tijdens een acute vaso-occlusieve crisis (VOC), niet zelden in combinatie met ACS of orgaanfalen. Acuut nierfalen kan ook een gevolg zijn van het blootstellen van patiënten met chronische sikkelcelnefropathie of andere nieraandoeningen aan nefrotoxische geneesmiddelen zoals NSAID’s of intraveneuze contrastvloeistof.
4.7 Cardiopulmonaire aandoeningen
Liefst 20 tot 48 procent van de kinderen met sikkelcelziekte krijgt te maken met complicaties aan de longen en luchtwegen: acute chest syndrome, astma, recurrente wheezing zonder astma, pulmonale hypertensie, acute en chronische veneuze trombo-embolie, pulmonale fibrose en
11 obstructief slaapapneusyndroom. Respiratoire insufficiëntie manifesteert zich meestal pas op volwassen leeftijd en is vaak het gevolg van één van bovenstaande complicaties. Ook het hart is kwetsbaar, met pulmonale hypertensie (PH) en cardiomegalie als belangrijke complicaties. 4.7.1 Acute Chest Syndrome
Bij acute chest syndrome (ACS) zorgt het sikkelen in de bloedvaten voor een verminderde toevoer van zuurstof naar de longen, waardoor longschade optreedt. ACS is een bijzonder ernstige complicatie van sikkelcelziekte: het is de tweede meest frequente reden voor hospitalisatie van kinderen en volwassenen met sikkelcelziekte en de meest voorkomende oorzaak van overlijden (9, 12). Meer dan 50% van de kinderen met sikkelcelziekte maken een ACS-episode door voor ze 10 jaar worden (26), met een piek tussen de leeftijd van 2 en 4. ACS kan mild of ernstig zijn, met mogelijke progressie tot respiratoir of multi-orgaanfalen. ACS wordt veroorzaakt door in situ sikkelen of embolieën door infarctering van het beenmerg (12). De symptomen van ACS zijn die van een lage-luchtweginfectie: thoracale pijn, hoesten, koorts, dyspneu en tachypneu. Kinderen hebben vaak koorts en de bovenste of middelste longkwab worden aangetast, in tegenstelling tot volwassen die vaak afebriel zijn en waarbij de ziekte multilobair is (10). ACS lijkt sterk op een longontsteking en kan zeer acuut en ongemerkt opzetten, tijdens hospitalisatie voor een VOC of na een chirurgische ingreep. Het is belangrijk ACS te onderscheiden van pneumonie om ongeschikte en inadequate behandeling te vermijden (27). Bloedanalyse kan een daling van de hemoglobineconcentratie tonen tot onder de baseline van de patiënt (10). Klinisch wordt ACS beschreven als de combinatie van symptomen van een lage-luchtweginfectie, koorts en nieuw longinfiltraat op beeldvorming (9, 12).
Bij patiënten met recurrente ACS-episodes, wordt (bij volwassen patiënten) occasioneel ook pulmonaire fibrose vastgesteld, maar in de pediatrische literatuur is hier weinig over te vinden.
4.7.2 Pulmonale Hypertensie
Pulmonale hypertensie (PH) wordt gedefinieerd als een verhoogde gemiddelde arteriële druk in rust van ≥ 25mm Hg ter hoogte van de arteria pulmonalis (mPAP) bepaald door katheterisatie van de rechter harthelft (RHC) (28). Ze wordt vaak geschat aan de hand van echocardiografie, met een snelheid van de tricuspidalis regurgitatie (TRV) ≥ 2.5 m/sec als surrogaat voor mPAP ≥ 25mm Hg, hoewel deze cut-off waarde enigszins controversieel is (29).
Symptomen zijn kortademigheid bij routine-activiteiten, vermoeidheid, lethargie, pijn op de borstkas, palpitaties, syncopes, perifeer oedeem en een verminderde eetlust (10). De complicatie ontstaat bij patiënten met sikkelcelziekte als gevolg van de chronische vasculaire
12 veranderingen ten gevolge van argininedepletie, verstoorde bio-beschikbaarheid van stikstofoxide en het vrijkomen van hemoglobine uit de rode bloedcellen (12).
Er is geen eensgezindheid over de prevalentie van PH bij kinderen met sikkelcelziekte. Prevalentie, niet-invasief bepaald (TRV≥ 2.5 m/s), verschilt tussen onderzoeken van 26 – 47% tot 8 – 11% (29). Meer recent onderzoek brengt het cijfer op 30%, over de genotypes heen (30). 4.7.3 Veneuze trombo-embolie
Ook de veneuze trombo-embolie (diep-veneuze trombose en pulmonale embolie) is een mogelijke complicatie. Risicofactoren en prevalentie werden recent bijgesteld, met prevalenties tussen 1.7% (2018, n=10.454)(31) en 2.9% (2018, n=414)(32), hoofdzakelijk bij kinderen met HbSS, met een centraal veneuze katheter als onafhankelijke risicofactor.
4.7.4 Respiratoire aandoeningen
Kinderen met sikkelcelziekte worden ook getroffen door andere respiratoire aandoeningen. Astma is een veel voorkomende complicatie, met een prevalentie tussen 8 en 53% (33). Astma verhoogt bovendien de kans op hospitalisatie voor een VOC of ACS (27, 34). De vernauwing van de luchtwegen bij astma kan bovendien leiden tot lokale hypoxie, het verder bevorderen van sikkelen van rode bloedcellen en systemische inflammatie (35). Patiënten met sikkelcelanemie én astma hebben een dubbel zo hoge mortaliteitskans, na correctie voor de gekende risicofactoren. Recurrent wheezing is vooral bij jonge kinderen een veelvuldige complicatie van sikkelcelziekte, ook zonder een diagnose van astma (27).
Kinderen met sikkelcelziekte hebben tevens een hoger risico op slaapproblemen (waaronder restless legs-syndroom, korte-termijn of chronische insomnie en sleep onset latency) en ademhalingsgebonden slaapstoornissen (sleep-disorderded breathing, SDB). Bij SDB is obstructief slaapapneusyndroom (OSAS) de meest prevalente: zo’n 41% van alle kinderen wordt erdoor getroffen, waarvan 10% met een ernstige variant (36). OSAS gaat gepaard met snurken, vermoeid ontwaken en slaperigheid overdag. Kinderen met sikkelcelziekte en OSAS hebben bovendien een hogere kans op hypoxemie, hypercapnie en acidose, wat het sikkelen van de erytrocyten kan bevorderen. Ademhalingsgebonden slaapstoornissen dragen mogelijk bij aan cardiopulmonaire complicaties bij kinderen met sikkelcelziekte (37).
4.8 Hepatobiliaire aandoeningen
Aandoeningen aan de galwegen (cholelithiasis, cholecystitis, galblaas sludge) zijn veelvoorkomende complicaties. Ze worden veroorzaakt door een verhoging van het ongeconjugeerd bilirubine, hyperbilirubinemie (10, 12). Bij kinderen is cholelithiasis, ten
13 gevolge van chronische hemolyse, de belangrijkste hepatobiliaire complicatie (12): bij 12% van de kinderen tussen 2 – 4 jaar worden bij een echo galstenen aangetroffen, een cijfer dat toeneemt tot 43% bij patiënten tussen 15 – 18 jaar (10). Patiënten zijn meestal asymptomatisch of presenteren zich met pijn in het rechterbovenste kwadrant van de buik of (milde) icterus (12). Ook de lever is kwetsbaar bij kinderen met sikkelcelziekte. Acute hepatische sequestratie en acute intrahepatische cholestase zijn bekende complicaties, voornamelijk bij patiënten met sikkelcelanemie. Leversequestratie wordt gekenmerkt door een vergroting van de lever en een daling van minstens 2g/dL tegenover de baseline Hb-waarden. Acute intrahepatische cholestase is een zeldzame, acute vorm van leverschade die optreedt wanneer de gesikkelde erytrocyten de bloedvaten in de lever afsnoeren, wat voorkomt dat zuurstof het leverweefsel bereikt. Leverschade kan ook optreden door een teveel aan ijzer bij frequente bloedtransfusies.
4.9 Genito-urinaire aandoeningen, ontwikkeling en seksualiteit
Jongens met sikkelcelziekte kunnen geconfronteerd worden met priapisme: ongewilde, langdurige, pijnlijke erecties doordat het bloed onvoldoende kan afvloeien uit de penis (38). Priapisme kent twee patronen:
• ‘Intermittent (stuttering) priapisme’: meerdere episodes korter dan 2 à 3 uur (12), die frequent plaats vinden. Kan ook een langdurige episode aankondigen.
• ‘Langdurig priapisme’: episode van ≥ 4 uur, vraagt onmiddellijk medisch ingrijpen. Liefst 80 procent van alle mannelijke patiënten met sikkelcelziekte, bij alle genotypes van de ziekte, worden geconfronteerd met priapisme (stuttering of langdurig) voor ze de leeftijd van 18 jaar bereiken, met zelfs gevallen van priapisme bij 3 tot 5-jarige jongetjes (38, 39).
Ook de groei en ontwikkeling kunnen hinder ondervinden, met name bij kinderen met sikkelcelanemie. Zij bereiken normaal gezien seksuele maturiteit, maar vaak met vertraging. Uit een recente literatuurstudie door Huang et al. (40) blijkt dat jongens met sikkelcelanemie de puberteit bereiken en meetbare verschillen vertonen in secundaire seksuele karakteristieken: minder gezichts- en lichaamsbeharing, later bereiken van de Tanner stadia, kleinere testikelgrootte en een kortere penislengte. Ze vertonen ook hormonale afwijkingen, zoals lage testosteronniveaus.
Ook adolescente meisjes met sikkelcelziekte worden geconfronteerd met gynaecologische uitdagingen, waaronder een vertraagde puberteit die gekenmerkt wordt door een latere start van de menarche, vaso-occlusieve pijn geassocieerd met de menstruele cyclus en onder-gediagnosticeerde, abnormale uteriene bloedingen (41).
14 4.10 Osteoarticulaire aandoeningen
Op osteoarticulair gebied zijn patiënten, veeleer adolescenten en volwassenen, met sikkelcelziekte kwetsbaar voor osteonecrose (avasculaire necrose), osteomyelitis en artritis. Avasculaire necrose (ook aseptische necrose, osteonecrose of ischemische necrose) is het afsterven van botweefsel door de aangetaste bloedtoevoer. Necrose ontstaat wanneer capillairen afgesloten worden door sikkelende erythrocyten in distale stukken van het bot, in de buurt van een gewricht waar de hypoxie maximaal is en de collaterale circulatie onvoldoende is (10). Bij kinderen is sikkelcelziekte de belangrijkste oorzaak van avasculaire necrose (42). De heup is het meest getroffen gewricht, gevolgd door de schouder. In onderzoek van Almeida-Matos et al. (2016, n=92)(43), werd een associatie gevonden met lage HbF, wat de theorie versterkt dat HbF ageert als een beschermende factor tegen avasculaire necrose (43).
Osteomyelitis is eveneens een veel voorkomend probleem bij patiënten met sikkelcelziekte, ook kinderen. Osteomyelitis wordt vaak getriggerd door een vaso-occlusieve crisis, maar kan ook worden veroorzaakt door een salmonella-infectie (42, 44).
4.11 Ulcus cruris
Sikkelcel ulcera (ulcus cruris) zijn ulcera die meestal klein starten maar snel groter worden. Ze ontwikkelen zich meestal op de benen en kunnen geïsoleerd of in groep voorkomen. Sommige van deze wonden genezen snel, anderen kennen een trage genezing en kunnen ook terugkomen. Kinderen worden meestal slechts vatbaar voor sikkelcel ulcera vanaf de leeftijd van 10 jaar. 4.12 Psychosociaal
De relatie tussen een chronische ziekte en sociaal-emotionele moeilijkheden bij kinderen is complex en wordt beïnvloed door een veelvoud aan factoren. Depressie, angststoornissen, slaapstoornissen en middelenmisbruik worden aangehaald als veelvoorkomende comorbiditeiten van sikkelcelziekte bij kinderen (45, 46). Zo kunnen de symptomen van depressie, dat bij kinderen een prevalentie heeft van 4 – 6% (47), zich al heel vroeg presenteren, gemiddeld vanaf 9 jaar (48). Ouderlijk catastroferen heeft hierop een negatieve invloed (49). Ook slaapstoornissen (46) komen frequent voor en worden soms zelfs als storender ervaren dan chronische pijn (50). Ze kunnen leiden tot verminderd cognitief functioneren en internaliserend gedrag (45, 51). Kinderen met sikkelcelziekte zijn ook at risk voor angststoornissen, een geschatte 6% van de pediatrische patiënten met sikkelcelziekte krijgt een psychiatrische diagnose, waarvan 22% gediagnosticeerd wordt met angststoornissen (52). De bovenvermelde psychosociale comorbiditeiten hebben ook vaak een lagere (hydroxyureum)therapietrouw tot gevolg, wat het verdere verloop van de ziekte opnieuw negatief beïnvloedt (53, 54).
15 4.13 Conclusie
Een overzicht van de hierboven besproken complicaties werd voorzien in tabel 1 (Appendix).
5 Preventie & Behandeling
Patiënten met sikkelcelziekte ervaren een breed spectrum aan complicaties. Hun impact kan beperkt worden door een goede follow-up en educatie van de pediatrische patiënt en zijn familie. Hieronder wordt een overzicht gegeven van de meest recente aanbevelingen voor de follow-up van kinderen met sikkelcelziekte, met aandacht voor preventie en management van de verschillende complicaties. Daarnaast is er ook aandacht voor enkele evidence based therapieën voor sikkelcelziekte, waaronder hydroxyureum en bloedtransfusies (12).
5.1 Eerste bezoek en opvolgbezoeken
Wanneer bij een kind sikkelcelziekte wordt vastgesteld, moet het onmiddellijk doorverwezen worden naar een gespecialiseerde kinderarts of een interdisciplinair team dat ervaring heeft in de behandeling van kinderen met sikkelcelziekte. De gespecialiseerde arts kan een pediatrisch hemato-oncoloog zijn, of een met de problematiek bekende pediater.
In het eerste bezoek, uitgevoerd door een specialist, moet naast de klinische onderzoeken aandacht zijn voor educatie over veelvoorkomende complicaties van sikkelcelziekte bij zuigelingen, zoals koorts, dactylitis en miltsequestratie en het herkennen van symptomen die vragen om onmiddellijk medisch ingrijpen (12). Ook moet de familie worden uitgelegd hoe een miltpalpatie kan worden uitgevoerd en moet dit indien nodig regelmatig worden herhaald. Daarnaast moet in een eerste bezoek, en herhaald wanneer nodig tijdens opvolgbezoeken, genetisch advies worden verleend over de ziekte. Er moet onder andere worden gekaderd dat beide ouders een abnormaal hemoglobine-gen hebben, en dat een kans bestaat van 25 procent bij elke zwangerschap dat het kind geboren wordt met sikkelcelziekte. Ouders moeten worden aangemoedigd om deze informatie te delen met alle toekomstige partners, zodat alle risico’s gekend zijn bij eender welke volgende zwangerschap (55).
Het is belangrijk dat het kind ook daarna op regelmatige tijdstippen gevolgd wordt bij een specialist. Deze bezoeken duren idealiter minstens 20 – 30 minuten en zijn voorbehouden voor specifiek advies met betrekking tot de mogelijke complicaties van sikkelcelziekte, alsook specifieke vaccinatienoden verwant met sikkelcelziekte, evaluatie van ziekte-specifieke behandelingen zoals transfusie- en hydroxyureumtherapie en evaluatie van andere mogelijke ziekte-specifieke behandelingen. Op latere leeftijd moet er ook aandacht zijn voor
16 communicatie met de school van het kind rond het belang van hydratatie en het herkennen van symptomen die vragen om medisch ingrijpen.
Kinderen met ernstige sikkelcel ziekte (HbSS en HbS-β0-thalassemie) worden best elke twee maanden opgevolgd tijdens levensmaand 0 – 6, om de drie maanden tijdens maand 7 – 18 en daarna om de zes maanden. De mildere vormen (oa. HbSC en HbS-β+-thalassemie genotypes) kan deze frequentie gehalveerd worden (56).
Bij de bezoeken moeten minstens volgende onderzoeken routinematig worden uitgevoerd: • een klinisch onderzoek, met o.a. aandacht voor palpatie van de milt en lever (in
steady-state) en aandacht voor respiratoire problemen (astma, slaapapneu, …)
• een volledig bloedbeeld moet minstens jaarlijks worden uitgevoerd en de resultaten moeten aan de ouders worden bezorgd, zodat die bij medische noodgevallen in een extern ziekenhuis bij de hand zijn.
• lengte, gewicht en hoofdomtrek moeten worden gemeten, op de groeicurves worden uitgezet en vergeleken met eerdere waardes. Er kan een groeivertraging ontstaan tussen de leeftijd van 6 en 24 maanden, geassocieerd met een daling in hemoglobineniveaus, maar daarna moet de groei zich herstellen. Deze informatie is cruciaal om snel pubertaire vertragingen of een korte gestalte te kunnen beoordelen.
• bloeddruk moet gemeten worden bij elk bezoek, om zo snel hypertensie op te sporen. Door de verlaagde hemoglobinewaarden bij patiënten met sikkelcelziekte kan men een lagere bloeddruk verwachten, waardoor het belangrijk is een baseline te bepalen. Een voor de leeftijd hoger dan verwachte bloeddruk, moet altijd de aandacht trekken. • daarnaast moet ook de zuurstofsaturatie minstens jaarlijks worden gecontroleerd, met
behulp van een transcutane zuurstofsaturatiemeting. Deze zijn typisch lager bij patiënten met sikkelcelziekte, waardoor een baselinewaarde belangrijk is.
• typering van de bloedgroepantigenen, minstens voor het Rh-complex (CcDEe) en Kell-antigenen bij één van de eerste bezoeken, in anticipatie van een bloedtransfusie. Daarnaast moeten, in functie van leeftijd en mogelijke complicaties ook een aantal preventieve behandelingen en screenings worden geïnitieerd. Deze worden op basis van literatuuronderzoek uitvoerig besproken in de volgende paragrafen.
17 5.2 Screening en preventie van complicaties
5.2.1 Pijn
Een vaso-occlusieve crisis, die zich bij jonge kinderen presenteert als dactylitis, is de meest voorkomende reden voor hospitalisatie bij pediatrische patiënten met sikkelcelziekte. Het is daarom belangrijk ouders in te lichten over de verschillende mogelijkheden om hiermee om te gaan. Milde pijn kan thuis behandeld worden met warmte, afleiding, paracetamol, ibuprofen en orale opiaten – op basis van een op maat gemaakt pijnbestrijdingsplan (57). Ernstige pijn moet behandeld worden in een hospitalisatiesetting (10, 12, 58).
Belangrijk tijdens contactmomenten met de ouders is info te geven over de preventie van pijncrisissen. Uitlokkende factoren dienen zoveel mogelijk vermeden te worden. Hieronder vallen koude, hypoxie, infectie, stress en herhaalde inspanningen. Daarnaast moet er aangedrongen worden op een dagelijkse voldoende vochtinname (± 1.5 liter + 20ml / kg lichaamsgewicht boven 20kg) (59) per dag en op zoutrijke voeding bij koorts, warm weer of fysieke inspanning aangezien dehydratie een belangrijke trigger is van een VOC (58, 60). Er is tot op heden geen klinische evidentie voor geneesmiddelen ter preventie van een VOC (39), maar de frequentie ervan neemt wel af bij behandeling met ziekte-specifieke behandelingen zoals hydroxyureumtherapie (4) .
Ook chronische pijn, gedefinieerd als pijn die langer dan drie maanden aanhoudt, moet goed worden opgevolgd. Chronische problematiek bij pediatrische patiënten stelt zich voornamelijk bij adolescenten, wanneer typisch ook de ziekenhuisverblijven langer worden (57). De behandeling ervan is moeilijk gezien de vele factoren die hieraan bij kunnen dragen (12). Vaak is behandeling een combinatie van paracetamol, NSAID’s, oordeelkundig gebruik van opiaten, antidepressiva en een multidisciplinaire aanpak (kinesitherapie en gedragstherapie) (10, 12). In de National Heart, Lung, and Blood Institute (NHLBI)-richtlijnen wordt aangestuurd op minstens een jaarlijkse evaluatie van chronische pijn bij patiënten met sikkelcelziekte met daarbij een beschrijving van de pijn, ernst (numerieke schaal), plaats, factoren die de pijn versterken of verlichten en bio-psychosociale factoren zoals de gemoedstoestand van de patiënt, huidige activiteiten en levenskwaliteit (10, 57).
Tenslotte moet de omgeving van het kind ook worden ingelicht over het herkennen van pijn: • Bij zeer jonge kinderen is verminderde activiteit vaak een eerste teken. Er kan zich een
symmetrische of asymmetrische zwelling ontwikkelen in het dorsum van de handen of voeten (dactylitis). Pijn wordt al dan niet vergezeld door huilen van het kind.
18 • Bij schoolgaande kinderen zal een kind zich apathisch gedragen of de pijn proberen te
negeren. Dat kan escaleren naar moeilijk behandelbare, overweldigende pijn.
• Bij oudere kinderen is het belangrijk dat ze ingelicht worden over het vermijden van de verschillende triggers en de manieren om thuis (of op school, …) de pijn te bestrijden. 5.2.2 Koorts en infecties
Koorts en infecties worden voornamelijk preventief aangepakt met behulp van vaccinaties en profylactische inname van penicilline, een aanpak die geleid heeft tot een drastische daling in de mortaliteit van sikkelcelziekte (4).
Voor de vaccinaties is er consensus, zowel in de NHLBI-richtlijnen (10), het advies van de Hoge Gezondheidsraad (61), als in de recente literatuur (62, 63) dat patiënten naast de standaardvaccinaties ook gevaccineerd moeten worden tegen invasieve pneumokokken met het 13-valente conjugaat vaccin (PCV13) kort na de geboorte, gevolgd door een vaccinatie met het 23-valente polysaccharidevaccin (PPSV23) op 2-jarige leeftijd met een herhaling op 5-jarige leeftijd. Patiënten met HbSC hebben een betere vaccinatierespons dan patiënten met HbSS. Een slechte vaccinatierespons wordt geassocieerd met een hogere kans op pulmonaire complicaties (ACS, pneumonie) op latere leeftijd (64). Kinderen met sikkelcelziekte blijven desondanks een verhoogde kans hebben op invasieve pneumokokkenziekte, voornamelijk voor serotypes van de pneumokok waartegen PCV13 niet beschermt (62, 65).
Verder wordt het standaard vaccinatieschema van Kind & Gezin aangeraden, aangevuld met het jaarlijks influenzavaccin (63, 66-70), meningokokken (B, ACWY)(63, 71) en varicella (70, 72). Varicella is namelijk een gekende oorzaak van stroke bij kinderen, waarvoor kinderen met sikkelcelziekte al verhoogd at risk zijn (73, 74). Voor meningokokken (B, ACWY) is er het advies van de Hoge Gezondheidsraad voor personen met functionele asplenie, inclusief sikkelcelziekte (71). Alle bovenvermelde vaccinaties zijn ook te vinden in de richtlijnen van het CDC (Centers for Disease Control and Prevention) voor patiënten met functionele asplenie, inclusief sikkelcelziekte (72).
Het is daarnaast ook belangrijk de familie te mee te geven laagdrempelig medische hulp te zoeken bij koorts (> 38.5°), aangezien pneumokokkensepsis nooit helemaal uit te sluiten is. Koorts kan ook het gevolg kan zijn van een parvovirus B19-infectie, pyelonefritis, cholecystitis, osteomyelitis of ACS (12).
In de NHBLI-richtlijnen (11) wordt, op basis van de PROPS I-studie (13, 56), aangeraden om tweemaal daags profylactisch orale penicilline toe te dienen aan kinderen (125mg voor kinderen
19 jonger dan 3 jaar, 250mg voor kinderen ouder dan drie jaar) met HbSS. Een recente Cochrane-review bevestigt deze aanbeveling (75) voor alle patiënten met sikkelcelanemie (HbSS en HbS-β0-thalassemie). Deze kunnen veilig gestaakt worden van zodra het kind vijf jaar oud is (76),
tenzij het kind een splenectomie onderging of het alsnog een infectie met pneumokokken doormaakte. Het is belangrijk zeker te zijn dat het kind volledig gevaccineerd is tegen pneumokokken vooraleer het profylactisch toedienen van penicilline wordt stopgezet.
5.2.3 Cerebrovasculaire aandoeningen
Preventie en screening zijn belangrijk om patiënten met een verhoogde kans op een beroerte in kaart te brengen (12, 14). Routinescreening met transcraniële doppler ultrasonografie (TCD) in combinatie met bloedtransfusietherapie, vermindert het risico op een CVA tegen 18-jarige leeftijd bij patiënten met sikkelcelanemie van 18% naar 1.9% (2, 77, 78).
Er wordt vanuit de literatuur aanbevolen patiënten met sikkelcelanemie jaarlijks te screenen met TCD tussen 2 – 16 jaar (10, 12, 79, 80). Hiermee kan verhoogde transcraniële velocity, TCV, secundair aan stenose, worden gedetecteerd. Verhoogde TCV indiceert een verhoogd risico op een beroerte bij kinderen met sikkelcelanemie (79, 80). Kinderen metrandverhoogde (170 tot 199 cm per seconde) of verhoogde (≥ 200 cm per seconde) TCV-resultaten moeten worden doorverwezen naar een kinderhemato-oncoloog met ervaring in lange-termijn transfusietherapie, om zo de kans op een beroerte te doen dalen (11, 80). Effectiviteit van transfusietherapie, met als doel het HbS onder de 30% te houden, in de preventie van een CVA bij sikkelcelanemie-patiënten is bewezen in de STOP-trial. Wanneer de therapie wordt stopgezet, gaat het risico terug naar de baselinewaarde (81, 82).
Daarnaast moet ook gescreend worden op (de gevolgen van) een stil infarct. Verminderde schoolprestaties, een ontwikkelingsachterstand of gedragsstoornissen moeten dus bevraagd worden bij routine-evaluaties. Een positieve screening voor leer- en ontwikkelingsachterstand voorspelt een verhoogde kans op beroerte (83). Screening hiervan is eenvoudig, met vragen als “heeft uw kind problemen gehad op school, bijvoorbeeld met een academisch onderwerp, emotioneel, gedragsmatig of sociaal? Indien ja – leg uit aan de hand van kernwoorden uit acht categorieën: academische prestaties, aanwezigheidsproblemen, aandachtsproblemen, gedragsproblemen, motorische problemen, sociale problemen of spraakproblemen.”
Sommige experten adviseren te screenen op stille infarcten met behulp van een MRI-scan bij alle patiënten met sikkelcelanemie vanaf 5 jaar oud (12) en alle patiënten met sikkelcelziekte met verminderde schoolprestaties, een ontwikkelingsachterstand of gedragsstoornissen (10, 12). Bernaudin et al. (2014, n=189) (77) identificeerden ook extracraniële arteria carotis interna
20 stenose als een onafhankelijke risicofactor voor stille infarcten bij patiënten met sikkelcelanemie en raden daarom aan om de meting van extracraniële circulatie (via MRA) mee op te nemen in het protocol voor screening op stille infarcten (12, 77). Acute anemie en baseline hemoglobine niveaus < 7 g/dL werden ook geïdentificeerd als onafhankelijke risicofactoren voor een stil infarct (77). Therapieën die deze hemoglobineniveaus positief beïnvloeden (hydroxyureumtherapie, transfusietherapie) kunnen het risico op een stil infarct verkleinen (17, 84). Ook besmetting met parvovirus B19 is een risicofactor voor stille infarcten (17).
Voor asymptomatische kinderen met andere genotypes dan sikkelcelanemie wordt screening afgeraden in de NHLBI-richtlijnen (10, 11).
Het is verder de taak van de behandelende arts om de verzorgers en familie van het kind in te lichten over hoe ze symptomen van een CVA kunnen herkennen: het niet kunnen bewegen van een gezichtsdeel, arm of been zonder dat pijn hiervan aan de oorzaak ligt. Een stil infarct kan worden herkend aan leer- en gedragsstoornissen, of een ontwikkelingsachterstand.
5.2.4 Anemie
Om patiënten met anemische crisissen (aplastische crisis, miltsequestratie) goed te kunnen opvolgen is het belangrijk bij elk routineonderzoek een volledig bloedbeeld af te nemen, om zo een volledige geschiedenis te creëren, met duidelijke baselinewaarden. Ook de serologie van het parvovirus B19 (IgG, IgM), dat een aplastische crisis kan veroorzaken, moet daarbij worden meegenomen tot ze positief is. Er bestaat nog geen vaccin tegen parvovirus B19 (20).
Bij kinderen waar autoinfarctering van de milt nog niet heeft plaatsgevonden (typisch < 5 jaar bij HbSS en HbSβ0-thalassemie, later bij patiënten met HbSC en HbSβ+-thalassemie), bestaat het risico op miltsequestratie. Dit is de meest acute oorzaak van abdominale pijn bij sikkelcelziekte-patiënten (39) en wordt o.m. behandeld met een transfusie (10).
Daarom wordt de familie van de patiënten geleerd hoe ze de symptomen van ernstige anemie kunnen herkennen en hoe de milt gepalpeerd kan worden om splenomegalie herkennen (3, 12). Bij een routine-onderzoek word daarom ook de milt in steady-state geëvalueerd.
Bij patiënten met recurrente acute miltsequestratie of symptomatische hypersplenisme (waarbij de milt chronisch vergroot en deels necrotisch is, waardoor ze niet naar normale grootte kan terugkeren), wordt een splenectomie aanbevolen (21, 85). Verder onderzoek naar de meest geschikte leeftijd om dit uit te voeren is nodig, bovendien is dit niet complicatievrij (21, 56).
21 5.2.5 Oftalmologische aandoeningen
Patiënten met sikkelcelziekte zijn vatbaar voor proliferatieve retinopathie (PSR). Daarom wordt in de literatuur screening met fundoscopie aanbevolen voor alle patiënten vanaf 9 à 10 jaar (10-12, 55, 86). In de literatuur is geen duidelijkheid over de timing hiervan. Jaarlijkse (11, 55) of tweejaarlijkse controles (10-12) worden geadviseerd. Bij detectie van PSR kan worden
ingegrepen met laserfotocoagulatie of vitrectomie, bij een glasvochtbloeding (12). 5.2.6 Nefrologische aandoeningen
Vanuit de NHLBI-richtlijnen en de recente literatuur wordt jaarlijkse urine-analyse (microalbuminurie, proteïnurie) aangeraden vanaf de leeftijd van 10 jaar (10, 11, 55, 87). Patiënten met proteïnurie (> 300 mg per 24 uur), microalbuminurie (albumine/creatinine ratio 30-300mg/g) of verhoogde serum creatinine niveaus (> 0.7 mg/dL) moeten worden doorverwezen naar een nefroloog. De nood aan deze jaarlijkse urine-analyse werd na een retrospectief onderzoek opnieuw bevestigd door Speller-Brown et al. (2018, n=205) (88). Uit recent onderzoek (88, 89) blijkt leeftijd de belangrijkste predictor van microalbuminurie met een sterke associatie met proteïnurie, hematurie, leeftijd, gewicht en systolische bloeddruk. Hypertensie kan dus eveneens een rol spelen in de ontwikkeling van nefrologische aandoeningen op jonge leeftijd bij patiënten met sikkelcelziekte. Patiënten met sikkelcelanemie hebben een hogere kans op microalbuminurie. Hierin wordt ook aangegeven dat er verder onderzoek moet gebeuren naar het verlagen van de leeftijd voor screening op microalbuminurie, zonder hierbij een leeftijd te formuleren. Wel wordt aangeraden om bij routinecontroles aandacht te hebben voor de bloeddruk van de patiënt (age-based norms, 90e en 95e percentiel), aangezien deze indicatief kan zijn voor de ontwikkeling van nefrologische aandoeningen (88). Er zijn studies waarbij microalbuminurie al op een leeftijd van 4 jaar werd ontdekt, maar 7 jaar wordt beschouwd als de typische leeftijd waarop dit zich ontwikkelt bij kinderen (23). Van zodra dit kan worden waargenomen, is er echter reeds significante nierschade opgetreden (25). Ook de GFR is een belangrijk hulpmiddel in de management van nefrologische aandoeningen zoals sikkelcelnefropathie, maar de methode waarmee deze bepaald wordt, overschat deze meestal bij sikkelcelpatiënten. Een consistente meting hiervan, startend vanaf een baseline, kan dan helpen om verschillen waar te nemen (23).
Nieuw onderzoek (2019, n=70) identificeert cystatine C-serum en β-2 microglobuline als vroege, specifieke en sensitieve biomarkers voor het identificeren van glomerulaire en tubulaire dysfunctie bij kinderen met sikkelcelziekte, maar meer onderzoek hiernaar is nodig (90), zeker aangezien eerder onderzoek (2015, n=45) dit tegenspreekt (91).
22 Tenslotte moet bij gesprekken met de omgeving van de patiënt het belang van voldoende hydratie blijvend benadrukt worden: dagelijkse vochtinname van minimum 2.5 liter per dag en zoutrijke voeding bij koorts, warm weer of fysieke inspanning.
5.2.7 Cardiopulmonaire aandoeningen 5.2.7.1 Acute Chest Syndrome
De ouders of familie van de patiënt moeten worden ingelicht over de gevaren van acute chest syndrome (ACS) voor het kind 6 – 9 maanden oud is. Gezien de gevaren van de complicatie, die sterke gelijkenissen vertoont met een pneumonie, is het belangrijk dat de ouders weten dat de complicatie initieel moet worden opgevolgd in een medische setting met parenterale antibiotica. Er moet worden benadrukt dat er laagdrempelig medische hulp moet worden gezocht bij kortademigheid, alsook dat astma een belangrijke promotor is van ACS. Daarom moeten deze scherp aangepakt worden en (preventieve) behandelplannen strik worden gevolgd. Ook blootstelling aan tabaksrook moet ten alle koste worden vermeden.
Preventie van ACS bestaat voornamelijk uit behandeling van sikkelcelziekte, met bloedtransfusie-, of hydroxyureumtherapie, terwijl screening bestaat uit het aandachtig zijn voor signalen en symptomen van respiratoire aandoeningen tijdens routineonderzoeken (92). 5.2.7.2 Respiratoire aandoeningen
Arteta et al. (93) raden aan bij alle patiënten met sikkelcelanemie (HbSS, HbS-β0-thalassemie) volledige pulmonaire functietesten (spirometrie, plethysmografie en longdiffusie capaciteit) uit te voeren, zeker bij kinderen met een geschiedenis van astma, wheezing en verhogingen in de hemolytische markers. Anim et al. (33) volgen in die aanbeveling voor alle patiënten met herhaalde episodes van ACS of met ernstige of ongecontroleerde astma (12, 93). Een gelijkaardige aanbeveling is te vinden in het NHLBI-richtlijnen (10), die aanraden om bij de routinematige klinische onderzoeken aandacht te hebben voor signalen en symptomen van respiratoire problemen, zoals astma, restrictieve longziekte of obstructieve slaapapneu. Bij kinderen met deze symptomen, kunnen pulmonaire functietesten de oorzaak van het probleem identificeren. Een routine screening met longfunctietesten of slaapstudies wordt door het NHLBI afgeraden voor alle asymptomatische kinderen met sikkelcelziekte (11, 12).
Gezien het risico op obstructieve pulmonaire aandoeningen en hyperresponsiviteit van de luchtwegen, gaat Mehari et al. (37) hier tegen in. Zij raden aan om bij alle kinderen met sikkelcelziekte 1- tot 3-jaarlijkse pulmonaire functietesten uit te voeren in de vorm van spirometrie. Ook Greenough at al. (94) benadrukt een routinematige, jaarlijkse opvolging van
23 de longfunctie bij kinderen, aangezien een snelle afname van de longfunctie vooral bij zeer jonge kinderen plaatsvindt (94). Bij kinderen jonger dan 5 jaar is het vaak technisch niet mogelijk om een longfunctietest uit te voeren. Diagnose van astma wordt in deze leeftijdsgroep vaak gesteld op basis van anamnese en klinisch onderzoek, eventueel kan impuls-oscillometrie gebruikt worden (95). Bij oudere kinderen kan bijkomend voor spirometrie gekozen worden. Er moet tijdens routineonderzoeken ook aandacht zijn voor (routinematig) snurken bij kinderen, aangezien dit indicatief is voor obstructief slaap apneu syndroom, OSAS (37) – net als niet-herstellende slaap, nachtelijk gasping of choking, hypersomnolentie tijdens de dag en een verlaagde zuurstofsaturatie bij ontwaken (37, 96). Adenotonsillectomie kan bij het voorkomen van OSAS overwogen worden (97, 98), mits de juiste perioperatieve zorg.
Kinderen met astmasymptomen moeten behandeld worden volgens de bestaande astmarichtlijnen en laagdrempelig worden doorgestuurd naar een kinderpneumoloog. Effectief astmamanagement kan helpen om sikkelcelziekte-gerelateerde symptomen te voorkomen (99). Patiënten met astma die gehospitaliseerd worden voor een VOC moeten behandeld worden met bronchodilatoren om verdere escalatie van de astmasymptomen te voorkomen (99).
5.2.7.3 Pulmonale Hypertensie
Een routine screening hiervoor bij asymptomatische kinderen met echocardiogram wordt niet aan- of afgeraden in de NHLBI-richtlijnen wegens onvoldoende bewijs van effectiviteit (11, 12). Screening wordt in diezelfde richtlijnen wel aangeraden bij een klinisch vermoeden van PH, waarvan inspanningsintolerantie een belangrijke aanwijzing is. Routine screening met een Doppler-echocardiografie vindt zijn oorsprong in het verband tussen een verhoogde TRV en mortaliteit bij sikkelcelziekte. Bij kinderen is er een verband tussen TRV en morbiditeit, wat zich uit in een groter risico op verminderde inspanningscapaciteit, gemeten aan de hand van een gestandaardiseerde inspanningstest (100, 101).
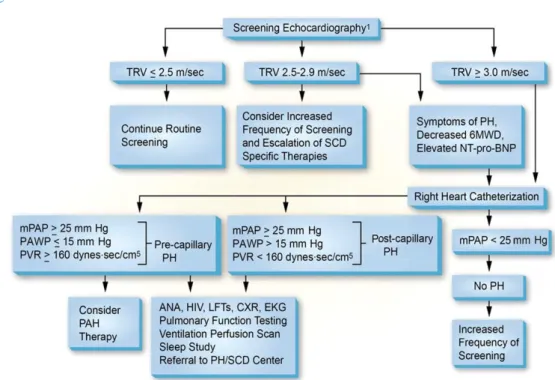
Tegelijk blijkt uit het quasi tegelijk gepubliceerde artikel van het American Thoracic Society’s Ad Hoc Committee on Pulmonary Hypertension of Sickle Cell Disease (101) dat zij “routinematig het risicoprofiel van de patiënt met sikkelcelziekte in kaart brengen door het TRV te meten aan de hand van een Doppler-echocardiografie”. Hieruit volgt aanbeveling om een patiënt elke 1 – 3 jaar te screenen met echocardiografieop basis van het protocol hieronder (fig. 1, zie Appendix), met een eerste screening op 8 jaar oud om een baseline TRV te bepalen (101, 102), vroeger bij patiënten met ernstige hemolytische anemie. Bij een TRV hoger dan 2.9m/s kan dan RHC worden uitgevoerd om de diagnose van PH vast te stellen.
24 Gordeuk et al. (103) geeft echter aan dat als RHC wordt uitgevoerd vanaf een TRV ≥ 2.9m/s, 4 op 10 patiënten met pulmonale hypertensie wordt gemist. Zij raden daarom een multimodale aanpak aan (voor volwassenen): uit hun onderzoek blijkt de combinatie van een TRV tussen 2.5m/s en 2.8m/s met een NT-proBNP waarde > 164.5pg/mL en een 6-minuten wandelafstand < 333m een positieve predictieve waarde te hebben van 62% met 7% vals-negatieven. Daaruit volgt de aanbeveling om een RHC uit te voeren bij een TRV tussen 2.5m/s en 2.8m/s, met een andere indicatie van PH zoals dyspneu, verhoogde NT-proBNP, hypertrofie van het rechterventrikel op een echocardiogram of beperkte inspannings- mogelijkheden – in lijn met het hierboven voorgestelde algoritme (101).
Deze algoritmes zijn echter bepaald voor volwassenen met sikkelcelziekte. De klinische significantie van een verhoogd TRV bij kinderen is minder goed gedefinieerd. Hebson et al. (2015, n=630)(104) en Liem et al. (2009, n=78)(105) konden een TRV ≥ 2.5m/s niet aanwijzen als een onafhankelijke predictor van PH. Gordeuk et al. (2011, n=160) (100) stelt vast dat TRV-waarden bij kinderen typisch hoger liggen, en de cut-off voor RHC daar hoger gesteld moet worden: TRV ≥ 2.7m/s (100). Lilje et al. (2017, n=91) stellen op basis van onderzoek een gemodificeerd protocol voor, met een cut-off TRV ≥ 2.9m/s voor bij kinderen.
Uit nieuw onderzoek van Yates et al. (2018, n=294)(30) blijkt vooral dat een verhoogd TRV bij kinderen sterk beïnvloed wordt door de mate van anemie en dat de hemoglobineconcentratie moet worden beschouwd als parameter voor initiatie van TRV-screening.
Bovenstaande resultaten duiden erop dat een verhoogde TRV risico kan voorspellen, maar er moeten tijdens een labo-onderzoek hogere cut-off waarden of bijkomende afwijkingen worden gevonden vooraleer over te gaan tot meer invasieve evaluaties. Preventieve screening voor PH bij kinderen vraagt dus verder onderzoek om correcte aanbevelingen te formuleren (102, 106). Voor kinderen met PH (RHC of TRV ≥ 2.5 m/sec), wordt hydroxyureumtherapie aangeraden of bij contra-indicatie hiervoor, chronische transfusietherapie.
Gezien de link tussen obstructief slaapapneu en PH, wordt ook een slaapstudie aanbevolen bij patiënten met een klinische diagnose van PH of TRV ≥ 2.5 m/sec (107).
5.2.7.4 Veneuze trombo-embolie
Over het langdurig gebruik van anticoagulatie bij kinderen met sikkelcelziekte en at risk voor een VTE (voornamelijk patiënten met HbSS), is weinig literatuur te vinden. Bij volwassenen is het gebruik geïndiceerd bij patiënten met PH, zonder gekend risico op bloedingen (101).
25 5.2.8 Hepatobiliaire aandoeningen
Belangrijkste aanwijzingen voor aandoeningen aan de galwegen bij een klinisch onderzoek zijn pijn in het rechter hypochondrium of milde icterus, al moet worden opgemerkt dat de meeste patiënten met galwegaandoeningen asymptomatisch zijn.
Bij patiënten met complicaties wordt een cholecystectomie aangeraden om verdere complicaties te vermijden (12). Onderzoek van Goodwin et al. (2016, n=191)(108) raadt aan om te screenen vanaf 8 jaar op afwijkende bloedresultaten (totaal bilirubine, AST, ….), en waar nodig electieve cholecystectomie te overwegen. Er wordt ook aanbevolen om beeldvorming voor niet-gerelateerde abdominale aandoeningen steeds uit te breiden naar een volledige echo abdomen, om zo vroeger complicaties op te sporen. Een protocol werd niet opgesteld.
Om vroegtijdig leveraandoeningen op te sporen is het belangrijk de grootte van de lever te bepalen in steady-state, alsook bij routineonderzoeken leverfunctietesten uit te voeren. Patiënten met sikkelcelanemie zijn vatbaarder en presenteren zich soms met directe hyperbilirubinemie zonder aanwijzing voor infectie of obstructie van de galwegen (12). Er is in de literatuur geen aanbeveling te vinden voor systematische screening met beeldvorming van asymptomatische patiënten (alle genotypes) op hepatobiliaire aandoeningen. 5.2.9 Genito-urinaire aandoeningen, ontwikkeling en seksualiteit
Het is belangrijk dat de omgeving van het kind ingelicht worden over deze complicatie voor de leeftijd van 5 jaar. Er wordt tijdens opvolgbezoeken een managementstrategie gecommuniceerd voor stuttering priapisme: fysieke inspanning, warme baden, urineren, ejaculatie, pijnstillers en verhoogde hydratie kunnen bijdragen aan detumescentie (12, 39). Daarnaast wordt ook het gevaar op permanente schade bij langdurig priapisme (> 4u) benadrukt. Dit is een urologische urgentie die moet worden behandeld om fibrose en impotentie te voorkomen.
Bij patiënten met een opvallend slechte groei en (seksuele) ontwikkeling, gekenmerkt door waarden onder het derde percentiel of bij kinderen waarvan de puberteit op 13 à 14-jarige leeftijd nog niet is gestart, is een doorverwijzing naar een kinderendocrinoloog aan te raden (109).
Bij adolescenten moet in de routineonderzoeken ook ruimte zijn voor educatie over seksualiteit en het risico op infertiliteit (110). Meisjes moeten worden ingelicht over de contraceptieve mogelijkheden. Hormonale contraceptiva kunnen menstrueel bloedverlies beperken, wat leidt tot hogere hemoglobineniveaus. Hormonale contraceptiva met enkel progestageen (pil, injecties en implantaten) hebben geen verhoogd risico op trombo-embolie en kunnen gebruikt worden
26 door vrouwen met sikkelcelziekte (10, 111). Een verleden van stroke is een contra-indicatie voor gecombineerde hormonale anticonceptie (oestrogeen en progestageen) (10).
5.2.10 Osteoarticulaire aandoeningen
Avasculaire necrose (AV) bij sikkelcelpatiënten uit zich voornamelijk in necrose van de femurkop. De NHLBI-richtlijnen raden aan om kinderen tijdens een klinisch onderzoek te evalueren op chronische of intermittente heuppijn. Avasculaire necrose wordt vaak pas in een laat stadium ontdekt (43), vroege screening kan dit vermijden. Bij klachten kan doorverwezen worden naar beeldvorming (röntgen, MRI) (10, 11). Hemoglobine - hematocriet ratio, als een proxy voor de gemiddelde corpusculaire hemoglobineconcentratie, is een beloftevolle nieuwe predictor voor osteonecrose bij kinderen met sikkelcelziekte (112).
Er is in de literatuur geen aanbeveling te vinden voor systematische screening met beeldvorming van asymptomatische patiënten (alle genotypes) op avasculaire necrose.
AV wordt behandeld met pijnstillers en kinesitherapie. Bij blijvende klachten volgt een doorverwijzing naar een orthopedisch chirurg voor een eventuele heupprothese (5, 23).
Preventie van osteoarticulaire aandoeningen bestaat voornamelijk uit ziekte-specifieke therapie (hydroxyureum, bloedtransfusies), het vermijden van triggers voor VOC en het vermijden van een besmetting met salmonella, aangezien deze kan leiden tot osteomyelitis.
5.2.11 Ulcus cruris
Als deel van het klinisch onderzoek bij een opvolgbezoek, moet de behandelende arts aandacht hebben voor de aanwezigheid van actieve of genezen ulcera op de onderste ledematen. Ulcera kunnen worden behandeld met een standaardbehandeling. Patiënten met therapieresistente diepe ulcera moeten worden geëvalueerd op osteomyelitis. Bij vermoeden van infectie of niet-genezende ulcera, moet een wondcultuur worden afgenomen en de mogelijke etiologieën onderzocht, om eventueel te behandelen met systemische of lokale antibiotica. Patiënten met therapieresistente ulcera worden doorverwezen naar een wondzorgspecialist (10, 11). Het belangrijkste aspect in het management van ulcus cruris is echter preventie. Traumapreventie, snelle behandeling van kleine wondjes, gebruik van insectenwerende middelen, het gebruik van steunkousen en hoogstand van de benen zijn hiervan de belangrijkste aspecten. De patiënt en zijn familie of verzorgers moeten hierover voldoende worden ingelicht (113).
5.2.12 Psychosociaal
Bij de routine-evaluatie van kinderen met sikkelcelziekte moet ook aandacht zijn voor symptomen van psychosociale comorbiditeiten zoals depressie en angststoornissen, vooral bij